|
| 1 | +# ProteinMPNN |
| 2 | +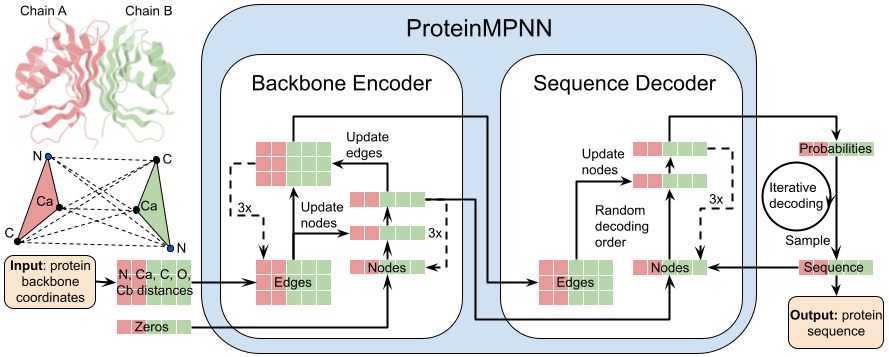 |
| 3 | +Read [ProteinMPNN paper](https://www.biorxiv.org/content/10.1101/2022.06.03.494563v1). |
| 4 | + |
| 5 | +To run ProteinMPNN clone this github repo and install Python>=3.0, PyTorch, Numpy. |
| 6 | + |
| 7 | +Full protein backbone models: `vanilla_model_weights/v_48_002.pt, v_48_010.pt, v_48_020.pt, v_48_030.pt`. |
| 8 | + |
| 9 | +CA only models: `ca_model_weights/v_48_002.pt, v_48_010.pt, v_48_020.pt`. Enable flag `--ca_only` to use these models. |
| 10 | + |
| 11 | +Helper scripts: `helper_scripts` - helper functions to parse PDBs, assign which chains to design, which residues to fix, adding AA bias, tying residues etc. |
| 12 | + |
| 13 | +Code organization: |
| 14 | +* `protein_mpnn_run.py` - the main script to initialialize and run the model. |
| 15 | +* `protein_mpnn_utils.py` - utility functions for the main script. |
| 16 | +* `examples/` - simple code examples. |
| 17 | +* `inputs/` - input PDB files for examples |
| 18 | +* `outputs/` - outputs from examples |
| 19 | +* `colab_notebooks/` - Google Colab examples |
| 20 | +* `training/` - code and data to retrain the model |
| 21 | +----------------------------------------------------------------------------------------------------- |
| 22 | +Input flags for `protein_mpnn_run.py`: |
| 23 | +``` |
| 24 | + argparser.add_argument("--ca_only", action="store_true", default=False, help="Parse CA-only structures and use CA-only models (default: false)") |
| 25 | + argparser.add_argument("--path_to_model_weights", type=str, default="", help="Path to model weights folder;") |
| 26 | + argparser.add_argument("--model_name", type=str, default="v_48_020", help="ProteinMPNN model name: v_48_002, v_48_010, v_48_020, v_48_030; v_48_010=version with 48 edges 0.10A noise") |
| 27 | + argparser.add_argument("--seed", type=int, default=0, help="If set to 0 then a random seed will be picked;") |
| 28 | + argparser.add_argument("--save_score", type=int, default=0, help="0 for False, 1 for True; save score=-log_prob to npy files") |
| 29 | + argparser.add_argument("--save_probs", type=int, default=0, help="0 for False, 1 for True; save MPNN predicted probabilites per position") |
| 30 | + argparser.add_argument("--score_only", type=int, default=0, help="0 for False, 1 for True; score input backbone-sequence pairs") |
| 31 | + argparser.add_argument("--conditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output conditional probabilities p(s_i given the rest of the sequence and backbone)") |
| 32 | + argparser.add_argument("--conditional_probs_only_backbone", type=int, default=0, help="0 for False, 1 for True; if true output conditional probabilities p(s_i given backbone)") |
| 33 | + argparser.add_argument("--unconditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output unconditional probabilities p(s_i given backbone) in one forward pass") |
| 34 | + argparser.add_argument("--backbone_noise", type=float, default=0.00, help="Standard deviation of Gaussian noise to add to backbone atoms") |
| 35 | + argparser.add_argument("--num_seq_per_target", type=int, default=1, help="Number of sequences to generate per target") |
| 36 | + argparser.add_argument("--batch_size", type=int, default=1, help="Batch size; can set higher for titan, quadro GPUs, reduce this if running out of GPU memory") |
| 37 | + argparser.add_argument("--max_length", type=int, default=200000, help="Max sequence length") |
| 38 | + argparser.add_argument("--sampling_temp", type=str, default="0.1", help="A string of temperatures, 0.2 0.25 0.5. Sampling temperature for amino acids. Suggested values 0.1, 0.15, 0.2, 0.25, 0.3. Higher values will lead to more diversity.") |
| 39 | + argparser.add_argument("--out_folder", type=str, help="Path to a folder to output sequences, e.g. /home/out/") |
| 40 | + argparser.add_argument("--pdb_path", type=str, default='', help="Path to a single PDB to be designed") |
| 41 | + argparser.add_argument("--pdb_path_chains", type=str, default='', help="Define which chains need to be designed for a single PDB ") |
| 42 | + argparser.add_argument("--jsonl_path", type=str, help="Path to a folder with parsed pdb into jsonl") |
| 43 | + argparser.add_argument("--chain_id_jsonl",type=str, default='', help="Path to a dictionary specifying which chains need to be designed and which ones are fixed, if not specied all chains will be designed.") |
| 44 | + argparser.add_argument("--fixed_positions_jsonl", type=str, default='', help="Path to a dictionary with fixed positions") |
| 45 | + argparser.add_argument("--omit_AAs", type=list, default='X', help="Specify which amino acids should be omitted in the generated sequence, e.g. 'AC' would omit alanine and cystine.") |
| 46 | + argparser.add_argument("--bias_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies AA composion bias if neededi, e.g. {A: -1.1, F: 0.7} would make A less likely and F more likely.") |
| 47 | + argparser.add_argument("--bias_by_res_jsonl", default='', help="Path to dictionary with per position bias.") |
| 48 | + argparser.add_argument("--omit_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies which amino acids need to be omited from design at specific chain indices") |
| 49 | + argparser.add_argument("--pssm_jsonl", type=str, default='', help="Path to a dictionary with pssm") |
| 50 | + argparser.add_argument("--pssm_multi", type=float, default=0.0, help="A value between [0.0, 1.0], 0.0 means do not use pssm, 1.0 ignore MPNN predictions") |
| 51 | + argparser.add_argument("--pssm_threshold", type=float, default=0.0, help="A value between -inf + inf to restric per position AAs") |
| 52 | + argparser.add_argument("--pssm_log_odds_flag", type=int, default=0, help="0 for False, 1 for True") |
| 53 | + argparser.add_argument("--pssm_bias_flag", type=int, default=0, help="0 for False, 1 for True") |
| 54 | + argparser.add_argument("--tied_positions_jsonl", type=str, default='', help="Path to a dictionary with tied positions") |
| 55 | +
|
| 56 | +``` |
| 57 | +----------------------------------------------------------------------------------------------------- |
| 58 | +For example to make a conda environment to run ProteinMPNN: |
| 59 | +* `conda create --name mlfold` - this creates conda environment called `mlfold` |
| 60 | +* `source activate mlfold` - this activate environment |
| 61 | +* `conda install pytorch torchvision torchaudio cudatoolkit=11.3 -c pytorch` - install pytorch following steps from https://pytorch.org/ |
| 62 | +----------------------------------------------------------------------------------------------------- |
| 63 | +These are provided `examples/`: |
| 64 | +* `submit_example_1.sh` - simple monomer example |
| 65 | +* `submit_example_2.sh` - simple multi-chain example |
| 66 | +* `submit_example_3.sh` - directly from the .pdb path |
| 67 | +* `submit_example_3_score_only.sh` - return score only (model's uncertainty) |
| 68 | +* `submit_example_4.sh` - fix some residue positions |
| 69 | +* `submit_example_4_non_fixed.sh` - specify which positions to design |
| 70 | +* `submit_example_5.sh` - tie some positions together (symmetry) |
| 71 | +* `submit_example_6.sh` - homooligomer example |
| 72 | +* `submit_example_7.sh` - return sequence unconditional probabilities (PSSM like) |
| 73 | +* `submit_example_8.sh` - add amino acid bias |
| 74 | +----------------------------------------------------------------------------------------------------- |
| 75 | +Output example: |
| 76 | +``` |
| 77 | +>3HTN, score=1.1705, global_score=1.2045, fixed_chains=['B'], designed_chains=['A', 'C'], model_name=v_48_020, git_hash=015ff820b9b5741ead6ba6795258f35a9c15e94b, seed=37 |
| 78 | +NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER/NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER |
| 79 | +>T=0.1, sample=1, score=0.7291, global_score=0.9330, seq_recovery=0.5736 |
| 80 | +NMYSYKKIGNKYIVSINNHTEIVKALKKFCEEKNIKSGSVNGIGSIGSVTLKFYNLETKEEELKTFNANFEISNLTGFISMHDNKVFLDLHITIGDENFSALAGHLVSAVVNGTCELIVEDFNELVSTKYNEELGLWLLDFEK/NMYSYKKIGNKYIVSINNHTDIVTAIKKFCEDKKIKSGTINGIGQVKEVTLEFRNFETGEKEEKTFKKQFTISNLTGFISTKDGKVFLDLHITFGDENFSALAGHLISAIVDGKCELIIEDYNEEINVKYNEELGLYLLDFNK |
| 81 | +>T=0.1, sample=2, score=0.7414, global_score=0.9355, seq_recovery=0.6075 |
| 82 | +NMYKYKKIGNKYIVSINNHTEIVKAIKEFCKEKNIKSGTINGIGQVGKVTLRFYNPETKEYTEKTFNDNFEISNLTGFISTYKNEVFLHLHITFGKSDFSALAGHLLSAIVNGICELIVEDFKENLSMKYDEKTGLYLLDFEK/NMYKYKKIGNKYVVSINNHTEIVEALKAFCEDKKIKSGTVNGIGQVSKVTLKFFNIETKESKEKTFNKNFEISNLTGFISEINGEVFLHLHITIGDENFSALAGHLLSAVVNGEAILIVEDYKEKVNRKYNEELGLNLLDFNL |
| 83 | +``` |
| 84 | +* `score` - average over residues that were designed negative log probability of sampled amino acids |
| 85 | +* `global score` - average over all residues in all chains negative log probability of sampled/fixed amino acids |
| 86 | +* `fixed_chains` - chains that were not designed (fixed) |
| 87 | +* `designed_chains` - chains that were redesigned |
| 88 | +* `model_name/CA_model_name` - model name that was used to generate results, e.g. `v_48_020` |
| 89 | +* `git_hash` - github version that was used to generate outputs |
| 90 | +* `seed` - random seed |
| 91 | +* `T=0.1` - temperature equal to 0.1 was used to sample sequences |
| 92 | +* `sample` - sequence sample number 1, 2, 3...etc |
| 93 | +----------------------------------------------------------------------------------------------------- |
| 94 | +``` |
| 95 | +@article{dauparas2022robust, |
| 96 | + title={Robust deep learning--based protein sequence design using ProteinMPNN}, |
| 97 | + author={Dauparas, Justas and Anishchenko, Ivan and Bennett, Nathaniel and Bai, Hua and Ragotte, Robert J and Milles, Lukas F and Wicky, Basile IM and Courbet, Alexis and de Haas, Rob J and Bethel, Neville and others}, |
| 98 | + journal={Science}, |
| 99 | + volume={378}, |
| 100 | + number={6615}, |
| 101 | + pages={49--56}, |
| 102 | + year={2022}, |
| 103 | + publisher={American Association for the Advancement of Science} |
| 104 | +} |
| 105 | +``` |
| 106 | +----------------------------------------------------------------------------------------------------- |
0 commit comments