Recommended File: MAF
Once you have a .maf multiple alignment, projected onto a reference genome, you can load this into GenomeView. Make sure that all contig ID’s used in the .maf file match the contig ID’s used in your genome sequence and annotation files.
If the annotations do not show up as a track, check your Configuration

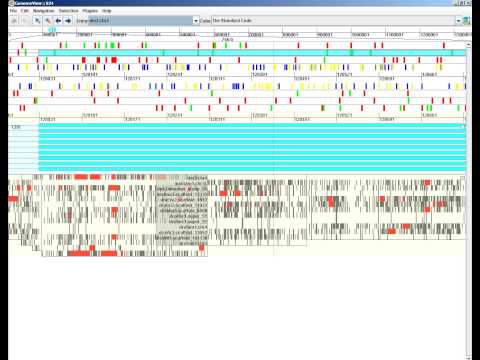
The Multiple Alignment Track visualizes alignments of many short-reads, showing how they align, and where mismatches are.
The following colors are used.
| Color | meaning |
|---|---|
| Gray | mismatch with reference |
| Red | gap in alignment |
| Green | Alignment to forward strand |
| Blue | Alignment to reverse strand |
| Yellow | Reference gap |
The visualization of multiple alignments depends on the zoom level.
When zoomed out very far, the track will not display alignments because of performance reasons.
Shows conservation plots. The alignments are displayed in gray. The more species align to a certain part of the reference sequence, the longer the gray line. Individual species are not displayed.

Shows conservation as a gray scale. Gaps in the alignment are displayed in red. On the middle level, we can still hover the track to see the species. An alignment on the forward strand is drawn in green, one to the reverse strand in blue.

Shows the individual nucleotides. Reference gaps are in yellow, alignment gaps in red . At the bottom of the track, the sequence logo for the aligment is shown. Mismatches and gaps are shown for each alignment. Hovering over the track displays the names of the species on the left.

You can follow our tutorial on downloading and using a MAF file.