018‐ scATAC‐Seq
Single cell ATAC sequencing with scRNA-Seq (scATAC-Seq)
library("iCellR")
my.data <- load10x("filtered_gene_bc_matrices/")
# see the row names
row.names(my.data)
# get peak names
ATAC <- grep("^chr",row.names(my.data),value=T)
# get scATAC data
MyATAC <- subset(my.data, row.names(my.data) %in% ATAC)
head(MyATAC)[1:3]
# AAACAGCCAAGTGAAC.1 AAACAGCCACTGACCG.1 AAACAGCCATGATTGT.1
#chr1.181218.181695 0 0 1
#chr1.191296.191699 0 0 0
#chr1.629770.630129 0 0 0
#chr1.633806.634251 0 0 0
#chr1.778422.779040 0 0 0
#chr1.827306.827702 0 0 0
dim(MyATAC)
# [1] 21923 6326
# get RNA data
MyRNAs <- subset(my.data, !row.names(my.data) %in% ATAC)
head(MyRNAs)[1:3]
# AAACAGCCAAGTGAAC.1 AAACAGCCACTGACCG.1 AAACAGCCATGATTGT.1
#MIR1302.2HG 0 0 0
#FAM138A 0 0 0
#OR4F5 0 0 0
#AL627309.1 0 0 0
#AL627309.3 0 0 0
#AL627309.2 0 0 0
dim(MyRNAs)
#[1] 36633 6326
# make iCellR object
my.obj <- make.obj(MyRNAs)
# add ATAC-Seq data
my.obj@atac.raw <- MyATAC
my.obj@atac.main <- MyATAC
# check your object
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 1.6.2
Raw/original data dimentions (rows,columns): 24127,6326
Data conditions: no conditions/single sample
Row names: MIR1302.2HG,TTLL10.AS1,MRPL20.AS1 ...
Columns names: AAACAGCCAAGTGAAC.1,AAACAGCCACTGACCG.1,AAACAGCCATGATTGT.1 ...
###################################
QC stats performed:FALSE, PCA performed:FALSE
Clustering performed:FALSE, Number of clusters:0
tSNE performed:FALSE, UMAP performed:FALSE, DiffMap performed:FALSE
Main data dimensions (rows,columns): 0,0
Normalization factors:,...
Imputed data dimensions (rows,columns):0,0
############## scVDJ-seq ###########
VDJ data dimentions (rows,columns):0,0
############## CITE-seq ############
ADT raw data dimensions (rows,columns):0,0
ADT main data dimensions (rows,columns):0,0
ADT columns names:...
ADT row names:...
############## scATAC-seq ############
ATAC raw data dimensions (rows,columns):21923,6326
ATAC main data dimensions (rows,columns):21923,6326
ATAC columns names:AAACAGCCAAGTGAAC.1...
ATAC row names:chr1.181218.181695...
############## Spatial ###########
Spatial data dimentions (rows,columns):0,0
########### iCellR object ##########
From here do the regular scRNA-seq as expleind above. See example below
# QC
my.obj <- qc.stats(my.obj,
s.phase.genes = s.phase,
g2m.phase.genes = g2m.phase)
# plot as mentioned above
# filter
my.obj <- cell.filter(my.obj,
min.mito = 0,
max.mito = 0.07 ,
min.genes = 500,
max.genes = 4000,
min.umis = 0,
max.umis = Inf)
# normalize RNA
my.obj <- norm.data(my.obj, norm.method = "ranked.glsf", top.rank = 500)
# normalize ADT
my.obj <- norm.adt(my.obj)
# gene stats
my.obj <- gene.stats(my.obj, which.data = "main.data")
# find genes for PCA
my.obj <- make.gene.model(my.obj, my.out.put = "data",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
no.cell.cycle = T,
out.name = "gene.model")
# run PCA and the rest is as above
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = my.obj@gene.model,data.type = "main")
# tSNE
my.obj <- run.pc.tsne(my.obj, dims = 1:10)
# UMAP
my.obj <- run.umap(my.obj, dims = 1:10)
# KNetL
my.obj <- run.knetl(my.obj, dims = 1:20, zoom = 200, dim.redux = "umap")
# clustering based on KNetL
my.obj <- iclust(my.obj, sensitivity = 200, data.type = "knetl")
# clustering based on PCA
# my.obj <- iclust(my.obj, sensitivity = 100, data.type = "pca", dims=1:10)
# check clusters and adjust if needed (optinal)
# cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
# my.obj <- change.clust(my.obj, change.clust = 3, to.clust = 4)
# my.obj <- change.clust(my.obj, change.clust = 3, to.clust = 10)
# order clusters
my.obj <- clust.ord(my.obj,top.rank = 500, how.to.order = "distance")
# plot
A= cluster.plot(my.obj,plot.type = "pca",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
B= cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
D= cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
library(gridExtra)
png('AllClusts.png', width = 12, height = 10, units = 'in', res = 300)
grid.arrange(A,B,C,D)
dev.off()
# save object
save(my.obj, file = "my.obj.Robj")
# find markers
marker.genes <- findMarkers(my.obj,
data.type = "main",
fold.change = 2,
padjval = 0.1,
uniq = F,
positive = T)
marker.genes1 <- cbind(row = rownames(marker.genes), marker.genes)
write.table((marker.genes1),file="marker.genes.tsv", sep="\t", row.names =F)
MyGenes <- top.markers(marker.genes, topde = 10, min.base.mean = 0.2, filt.ambig = F)
MyGenes <- unique(MyGenes)
png('heatmap_gg_genes.png', width = 10, height = 10, units = 'in', res = 300)
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters",cell.sort = F, conds.to.plot = NULL)
dev.off()Work on scATAC data (normalize and find marker peaks for each cluster)
# normalize ACAT
my.obj <- norm.data(my.obj, norm.method = "ranked.glsf", top.rank = 500, ATAC.data = TRUE, ATAC.filter = TRUE)
marker.peaks <- findMarkers(my.obj,
data.type = "atac",
fold.change = 2,
padjval = 0.1,
uniq = F,
positive = T)
marker.peaks1 <- cbind(row = rownames(marker.peaks), marker.peaks)
write.table((marker.peaks1),file="marker.peaks.tsv", sep="\t", row.names =F)
head(marker.peaks1)
# row baseMean baseSD
#chr17.64986035.64986113 chr17.64986035.64986113 0.01217359 0.18257818
#chr1.26542287.26542678 chr1.26542287.26542678 0.05828764 0.80077656
#chr4.8199063.8199275 chr4.8199063.8199275 0.04280424 0.56205649
#chr20.50274929.50275237 chr20.50274929.50275237 0.04684509 0.63361490
#chr2.218382038.218382236 chr2.218382038.218382236 0.03122394 0.31153105
#chr11.1760469.1760814 chr11.1760469.1760814 0.07050175 0.63322284
# AvExpInCluster AvExpInOtherClusters foldChange
#chr17.64986035.64986113 0.07868394 0.002346603 33.530999
#chr1.26542287.26542678 0.33849093 0.016887273 20.044144
#chr4.8199063.8199275 0.24803497 0.012481148 19.872769
#chr20.50274929.50275237 0.27007513 0.013862584 19.482308
#chr2.218382038.218382236 0.17736269 0.009631770 18.414340
#chr11.1760469.1760814 0.39043782 0.023230813 16.806894
# log2FoldChange pval padj clusters
#chr17.64986035.64986113 5.067424 1.187697e-05 2.875415e-02 1
#chr1.26542287.26542678 4.325109 3.653916e-05 8.539202e-02 1
#chr4.8199063.8199275 4.312721 6.059691e-06 1.489472e-02 1
#chr20.50274929.50275237 4.284093 2.871301e-05 6.779143e-02 1
#chr2.218382038.218382236 4.202758 6.359572e-10 1.677019e-06 1
#chr11.1760469.1760814 4.070981 6.813447e-11 1.800794e-07 1
# gene cluster_1 cluster_2
#chr17.64986035.64986113 chr17.64986035.64986113 0.0786839378 0.000000000
#chr1.26542287.26542678 chr1.26542287.26542678 0.3384909326 0.008895062
#chr4.8199063.8199275 chr4.8199063.8199275 0.2480349741 0.038672840
#chr20.50274929.50275237 chr20.50274929.50275237 0.2700751295 0.028703704
#chr2.218382038.218382236 chr2.218382038.218382236 0.1773626943 0.000000000
#chr11.1760469.1760814 chr11.1760469.1760814 0.3904378238 0.004537037
# cluster_3 cluster_4 cluster_5 cluster_6
#chr17.64986035.64986113 0.000000000 0.0000000000 0.0006934750 0.0010038760
#chr1.26542287.26542678 0.031485714 0.0029244992 0.0052261002 0.0041264535
#chr4.8199063.8199275 0.000000000 0.0007226502 0.0027450683 0.0113212209
#chr20.50274929.50275237 0.027092857 0.0121741140 0.0041820941 0.0051516473
#chr2.218382038.218382236 0.004292857 0.0042095532 0.0006722307 0.0038561047
#chr11.1760469.1760814 0.071678571 0.0177288136 0.0141820941 0.0061099806
# cluster_7 cluster_8
#chr17.64986035.64986113 0.003099029 0.009080357
#chr1.26542287.26542678 0.047110680 0.044484375
#chr4.8199063.8199275 0.014819417 0.028651786
#chr20.50274929.50275237 0.025499029 0.028765625
#chr2.218382038.218382236 0.011988350 0.035508929
#chr11.1760469.1760814 0.042093204 0.050708705
MyGenes <- top.markers(marker.peaks, topde = 10, min.base.mean = 0.2, filt.ambig = F)
MyGenes <- unique(MyGenes)
png('heatmap_gg_peaks.png', width = 10, height = 10, units = 'in', res = 300)
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters",cell.sort = F, conds.to.plot = NULL, data.type = "atac")
dev.off()
my.obj <- run.impute(my.obj,data.type = "knetl", nn = 10, ATAC.data = FALSE)
png('heatmap_gg_peaks.png', width = 10, height = 10, units = 'in', res = 300)
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters",cell.sort = F, conds.to.plot = NULL, data.type = "atac.imputed")
dev.off()
## you can also find avarage peak intensity per cluster
my.obj <- clust.avg.exp(my.obj, data.type = "atac")
head(my.obj@clust.avg)
#gene cluster_1 cluster_2 ...
#chr1.100037799.100038931 0.38238731 0.36750000 ...
#chr1.100132733.100133298 0.11195725 1.13593827 ...
#chr1.100249637.100250160 0.09851425 0.09511728 ...
#chr1.100265992.100266479 0.06768394 0.17707407 ...
#chr1.10032488.10033387 0.35273705 0.14885802 ...
#chr1.100352150.100352921 0.12006088 0.00000000 ...
# find out which cluster has the highest number
dat <- as.data.frame(t((my.obj@clust.avg)[,-1]))
dat <- hto.anno(hto.data = dat)
head(dat$assignment.annotatio)
#[1] cluster_1 cluster_2 cluster_4 cluster_2 cluster_3 cluster_4
#8 Levels: cluster_1 cluster_2 cluster_3 cluster_4 cluster_5 ... cluster_8Peak analysis
# make a bed file per cluster from the marker.peaks file you made up here
make.bed(marker.peaks)
# load packages
library(ChIPseeker)
library(clusterProfiler)
# load genome
require(TxDb.Hsapiens.UCSC.hg38.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene
Anno="org.Hs.eg.db"
# load bed files
Mylist1 = list.files(pattern=".bed")
Mylist1
Mylist <- as.list(Mylist1)
NAMES <- gsub('_peaks.bed','',Mylist1)
names(Mylist) <- NAMES
files <- Mylist
files
# perform analysis (example)
promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
tagMatrixList <- lapply(files, getTagMatrix, windows=promoter)
pdf("Plot_ProfileLineAll.pdf")
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000))
dev.off()
pdf('Plot_ProfileLine.pdf', width = 8, height = 10)
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000), facet="row")
dev.off()
pdf("Plot_heatmaps.pdf", width = 50, height = 6)
tagHeatmap(tagMatrixList, xlim=c(-3000, 3000), color=NULL)
dev.off()
# annotate
peakAnnoList <- lapply(files, annotatePeak, TxDb=txdb,
tssRegion=c(-3000, 3000), verbose=FALSE)
# plot annotatin
pdf("Plot_AnnoBar.pdf")
plotAnnoBar(peakAnnoList)
dev.off()
############### peak annotation
peakAnnoList <- lapply(files, annotatePeak, TxDb=txdb,
tssRegion=c(-3000, 3000), verbose=FALSE, annoDb=Anno)
capture.output(peakAnnoList, file = "peakAnnoList.txt")
genes = lapply(peakAnnoList, function(i) as.data.frame(i))
lapply(1:length(genes), function(i) write.table(genes[[i]],
file = paste0(names(genes[i]), ".xls"),
row.names = FALSE, sep="\t"))
Merging scATAC files with different intervals (as dipicted in bedtools website)
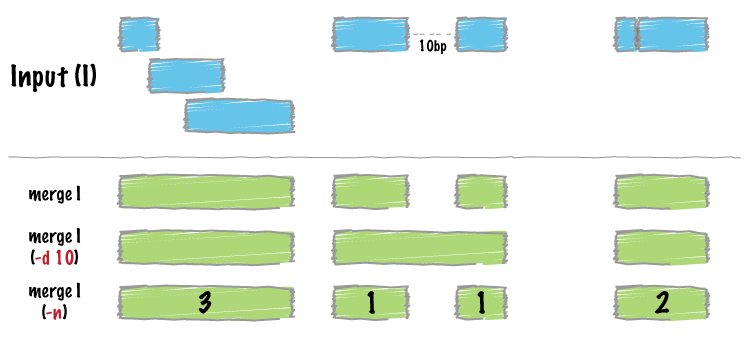
# Let's say you have 2 files that you need to merege
# example file
C <- load10x("count-JJsn_C_cDNA/",gene.name = 2)
M <- load10x("count-JJsn_M_cDNA/",gene.name = 2)
ATAC.C <- grep("^chr",row.names(C),value=T)
ATAC.M <- grep("^chr",row.names(M),value=T)
MyATAC.C <- subset(C, row.names(C) %in% ATAC.C)
MyATAC.M <- subset(M, row.names(M) %in% ATAC.M)
head(MyATAC.C)[1:3]
C <- MyATAC.C
M <- MyATAC.M
dim(C)
#[1] 58678 4211
dim(M)
#[1] 57776 4736
f1 <- row.names(C)
f2 <- row.names(M)
all.peaks <- c(f1,f2)
length(all.peaks)
#[1] 116454
# make a bed file
chr <- as.character(as.matrix(data.frame(do.call('rbind', strsplit(as.character(all.peaks),'.',fixed=TRUE)))[1]))
start <- data.frame(do.call('rbind', strsplit(as.character(all.peaks),'.',fixed=TRUE)))[2]
end <- data.frame(do.call('rbind', strsplit(as.character(all.peaks),'.',fixed=TRUE)))[3]
DAT <- as.data.frame(chr)
DAT$start <- as.numeric(as.matrix(start))
DAT$end <- as.numeric(as.matrix(end))
head(DAT)
# chr start end
#1 chr1 181218 181695
#2 chr1 191296 191699
#3 chr1 629770 630129
#4 chr1 633806 634251
#5 chr1 778422 779040
#6 chr1 827306 827702
# make Genomic Ranges
library("GenomicRanges")
all.gr <- GRanges(seqnames=DAT$chr,ranges=IRanges(start=DAT$start,end=DAT$end))
all.gr
#GRanges object with ?? ranges and 0 metadata columns:
# seqnames ranges strand
# <Rle> <IRanges> <Rle>
# [1] chr1 181218-181695 *
# [2] chr1 191296-191699 *
# [3] chr1 629770-630129 *
# [4] chr1 633806-634251 *
# [5] chr1 778422-779040 *
# ... ... ... ...
# [52] chr1 1303892-1306216 *
# [53] chr1 1307242-1309359 *
# [54] chr1 1324425-1325236 *
# [55] chr1 1348940-1349958 *
# [56] chr1 1372031-1372220 *
# -------
# seqinfo: 1 sequence from an unspecified genome; no seqlengths
################## sort and merge the peaks
mrg <- reduce(all.gr)
#Before merge
length(all.gr)
#[1] 116454
#after merge
length(mrg)
#[1] 71426
########################## choose file and give name
MyFile <- f1
name="f1_new.bed"
########################## copy paste the code here to make a new bed file
########################## the new bed has the old and new intervals (new intervals to be replaced with old)
chr <- as.character(as.matrix(data.frame(do.call('rbind', strsplit(as.character(MyFile),'.',fixed=TRUE)))[1]))
start <- data.frame(do.call('rbind', strsplit(as.character(MyFile),'.',fixed=TRUE)))[2]
end <- data.frame(do.call('rbind', strsplit(as.character(MyFile),'.',fixed=TRUE)))[3]
# make a bed file
DAT <- as.data.frame(chr)
DAT$start <- as.numeric(as.matrix(start))
DAT$end <- as.numeric(as.matrix(end))
MyFile <- DAT
# make intrval file to replace to new regions
MyFile.gr <- GRanges(seqnames=MyFile$chr,ranges=IRanges(start=MyFile$start,end=MyFile$end))
OverLap <- findOverlaps(MyFile.gr, mrg)
OLD1 <- (OverLap@from)
NEW1 <- (OverLap@to)
OLD = MyFile.gr[OLD1]
NEW = mrg[NEW1]
chr <- as.character(OLD@seqnames)
DAT <- as.data.frame(chr)
DAT$start <- OLD@ranges@start
DAT$end <- (OLD@ranges@start + OLD@ranges@width) - 1
DAT$new.chr<- as.character(NEW@seqnames)
DAT$new.start <- NEW@ranges@start
DAT$new.end <- (NEW@ranges@start + NEW@ranges@width) - 1
head(DAT)
# chr start end new.chr new.start new.end
#1 chr1 3247563 3248453 chr1 3247563 3248453
#2 chr1 3360706 3361554 chr1 3360706 3361554
#3 chr1 3552372 3553230 chr1 3552372 3553230
#4 chr1 3645171 3646034 chr1 3645093 3646034
#5 chr1 3670318 3671081 chr1 3670318 3671090
#6 chr1 3671326 3672230 chr1 3671314 3672230
dim(DAT)
#58678 6
length(MyFile.gr)
#58678
# diff
have = mrg[unique(NEW1)]
dontHave = mrg[-unique(NEW1)]
ADD <- dontHave
L <- length(as.character(ADD@seqnames))
chr <-rep("NA",L)
DAT1 <- as.data.frame(chr)
DAT1$start <- rep("NA",L)
DAT1$end <- rep("NA",L)
DAT1$new.chr<- as.character(ADD@seqnames)
DAT1$new.start <- ADD@ranges@start
DAT1$new.end <- (ADD@ranges@start + ADD@ranges@width) - 1
Final.DAT <- rbind(DAT,DAT1)
##### Write
write.table(Final.DAT,name,row.names=FALSE,sep="\t", quote = FALSE)
### reapeat this process for f2 (M) as well
# The first 3 columns are the original peaks and the last 3 are the ones that need to be replaced with original one. The NA peaks would also get the new peak ids but in the matrix the cells will have 0 for expressions. To do this use the iCellR function replace.peak.id.
MyATAC.C <- replace.peak.id(atac.data=MyATAC.C, bed.file = Final.DAT.C)
MyATAC.M <- replace.peak.id(atac.data=MyATAC.M, bed.file = Final.DAT.M)
# finally aggregate the samples and add to iCellR object
my.atac.data <- data.aggregation(samples = c("MyATAC1","MyATAC2","MyATAC3"),
condition.names = c("WT","KO","Ctrl"))
# add ATAC-Seq data
my.obj@atac.raw <- my.atac.data
my.obj@atac.main <- my.atac.data