双光子吸收是一种多光子吸收过程,与常见的一光子吸收(One-Photon Absorption, OPA)不同,TPA是指分子在极高光子通量密度下,同时吸收两个(或更多)能量较低的光子,从基态跃迁到激发态的物理过程 。
其基本原理可以概括为:
-
非线性效应:TPA是一种典型的非线性光学现象。在一光子吸收中,分子吸收一个能量等于或大于其电子跃迁能隙的光子。而在TPA中,分子通过一个虚拟中间态,同时吸收两个光子,这两个光子的能量之和等于分子从基态到激发态的跃迁能量。“虚拟态”并非真实的量子态,而是介于基态和最终激发态之间的一种瞬态,其寿命极短,由激光的强电场诱导产生。
-
激发条件:TPA要求极高的瞬时光子密度,通常需要使用飞秒(femtosecond, fs)或皮秒(picosecond, ps)脉冲激光器提供高峰值功率 。这是因为两个光子必须在极短的时间内(通常在 10−1610−16 秒以内)几乎同时与分子相互作用,才能发生协同吸收。
-
能量匹配:虽然单个光子的能量不足以引起跃迁,但两个光子的能量之和 $$ E=hν1+hν2 $$
必须等于或接近分子从基态$S_{0}$到某一激发态$S_n$的能级差。通常,这两个光子具有相同的能量(例如,两个近红外光子),因此所需的激发波长大约是对应一光子吸收波长的两倍。
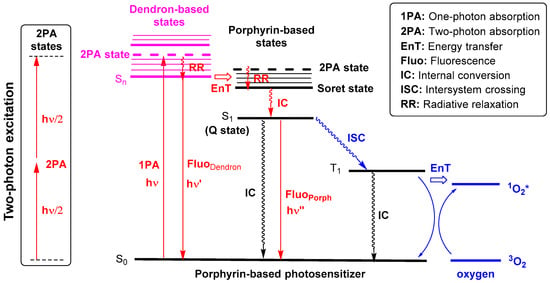
上图展示了光物理过程示意图。其中,1PA(一光子吸收)通过吸收一个光子将分子从基态(
$S_0$ )激发到第一激发单重态($S_1$ )。2PA(双光子吸收)则通过同时吸收两个光子将分子从基态($S_0$ )激发到更高的2PA激发态。这些激发态随后可以通过内部转换(IC)和系间窜越(ISC)等非辐射过程,或通过荧光(Flu)等辐射过程进行能量耗散。当分子通过ISC达到三重态($T_1$ )时,它可以与基态氧分子($^3O_2$ )相互作用,生成高活性的单线态氧($^1O_2$ ),这正是光动力治疗的关键机制 。 -
TPA截面(TPA Cross-section,
$δ$ ):衡量分子TPA效率的关键参数是TPA截面,单位通常为GM(Goppert-Mayer),其中$1 , \text{GM} = 10^{-50} , \text{cm}^4 \cdot \text{s} \cdot \text{photon}^{-1} \cdot \text{molecule}^{-1}$ 。高TPA截面是高效TPA材料设计的核心目标。一些研究已经报告了具有高TPA截面的材料,例如,一项研究设计了一种小分子吲哚方酸染料,其在780 nm处的TPA截面$ δ>8000 GM$。
传统的TPA材料设计和筛选高度依赖实验合成与表征,这是一个耗时且成本高昂的过程 。为了加速这一进程,研究者们转向计算方法和机器学习模型进行预测和虚拟筛选。
传统方法:基于化学知识的特征工程 早期和目前主流的TPA预测模型多采用“特征工程”的方法。这意味着研究人员会根据已有的化学直觉和物理化学原理,手动设计和提取分子的描述符(即特征)。这些特征通常具有明确的物理意义,例如:
- 共轭体系长度:TPA材料通常具有大的共轭体系,因为这有助于实现电荷转移,从而增强TPA响应 。
- 电子供体-受体(D-A)结构:许多高效TPA分子采用D-π-A(供体-π桥-受体)、D-π-D或A-π-A等构型。供体和受体基团的引入增强了分子的电荷转移特性,从而显著提高了TPA截面 。例如,含有三苯胺基团(TPA,作为强电子供体)的分子被广泛研究,其衍生物在有机发光二极管中表现出高发光效率,并影响吸收光谱和光致发光行为 。
- 溶剂极性:溶剂的极性也会显著影响TPA性能。一些研究发现,在极性溶剂中,分子的TPA截面会增加 ,而另一些研究则揭示溶剂效应对TPA光谱轮廓和强度有强烈影响,但对一光子吸收影响较小 。这表明溶剂环境对非线性光学过程的重要性。
- 分子对称性、几何构型:分子的对称性对TPA也有重要影响。例如,多核金属配合物可以结合多个TPA活性有机配体,形成复杂的多极体系,从而增强TPA效应,同时表现出更好的稳定性、更长的发射寿命和优化的光谱范围。
现有方法的局限性与新方法的探索 尽管基于化学知识的特征工程取得了成功,但其存在一些固有限制:
- 人工依赖性:特征的提取依赖于领域专家的经验和直觉,可能无法捕捉到分子中所有复杂的、非线性的相互作用。
- 特征完备性:手动设计的特征可能无法完全覆盖决定TPA行为的所有结构-性质关系。
- 泛化能力:基于特定类型分子设计的特征可能难以泛化到结构差异较大的新型TPA材料上。
为了克服这些挑战,本研究的动机是探索一种更为“端到端”的TPA预测模型,即通过更高级、更自动化的分子表示方法来捕捉分子结构中的复杂信息。具体而言,研究者尝试引入UniMol预训练模型。
UniMol预训练模型 UniMol是一种基于Transformer架构的分子表示学习模型,它通过在大规模分子数据集上进行预训练,学习到分子三维结构和化学键合的丰富信息。与传统的基于化学指纹或描述符的特征工程不同,UniMol能够生成高维、上下文敏感的分子表示(即“分子表示”或“分子嵌入”),这些表示被认为能更全面地编码分子的化学性质和结构信息。
通过结合UniMol生成的分子表示和已知的溶剂极性数据,研究旨在:
- 减少人工特征工程的依赖:直接利用预训练模型学习到的分子特征,避免了手动设计复杂化学描述符的过程。
- 捕捉更深层次的结构-性质关系:UniMol可能捕捉到传统特征工程难以发现的细微结构变化对TPA性能的影响。
- 构建端到端的预测模型:将分子结构直接映射到TPA截面值,简化了预测流程。
这种方法有望为TPA材料的理性设计提供更高效、更准确的预测工具,从而加速其在生物成像、光动力治疗等前沿领域的应用。同时,这也是机器学习在材料科学和化学领域应用的一个重要方向,即从传统的基于规则和特征工程的方法,向基于深度学习和表示学习的范式转变。
# UniMol特征提取
unimol_repr_extractor = UniMolRepr(data_type='molecule', remove_hs=False)
unimol_reprs = unimol_repr_extractor.get_repr(smiles_list, return_atomic_reprs=False)本研究采用UniMol预训练模型从SMILES字符串中提取512维的分子表示,随后通过PCA降维至100维,保留97.02%的方差信息。这种方法的优势在于无需依赖人工设计的化学描述符,而是通过大规模预训练获得的通用分子表示。
将UniMol分子表示与溶剂极性参数(Et30值)进行融合,构建101维的综合特征向量:
X = np.column_stack((cls_reprs_final, tpa_df['Et30'].values))针对TPA数据的极端偏态分布(方差远大于均值),采用对数变换进行数据标准化:
y_log = np.log1p(y) # 对数变换
scaler_y = StandardScaler()
y_log_scaled = scaler_y.fit_transform(y_log.reshape(-1, 1)).flatten()为解决参数过多导致的过拟合问题,设计了轻量化的神经网络架构:
class UltraSimpleTPANet(nn.Module):
def __init__(self, input_size):
super(UltraSimpleTPANet, self).__init__()
self.layer1 = nn.Linear(input_size, 32) # 101 → 32
self.dropout1 = nn.Dropout(0.3)
self.layer2 = nn.Linear(32, 16) # 32 → 16
self.dropout2 = nn.Dropout(0.2)
self.output_layer = nn.Linear(16, 1) # 16 → 1该模型将参数数量从68,225个大幅降至3,809个,参数/样本比例从110:1优化至6:1,有效缓解过拟合风险。
在测试集上的表现:
- MSE: 2,065,318.57
- RMSE: 1,437.12
- MAE: 715.74
- R²(原始空间): -0.018
- R²(对数空间): 0.291
通过超参数优化比较了多种机器学习算法:
- CatBoost: 验证RMSE 0.8505(最佳)
- XGBoost: 验证RMSE 0.8567
- LightGBM: 验证RMSE 0.8602
- Random Forest: 验证RMSE 0.8618
- SVR: 验证RMSE 0.8615
- ElasticNet: 验证RMSE 0.8732
-
数据特征: TPA数据呈现严重的长尾分布,原始数据方差(3,637)远大于均值(1,349),对数变换后分布更加合理。
-
模型复杂度: 初始模型参数过多导致严重过拟合,简化后的模型在保持预测能力的同时显著提升了泛化性能。
-
预测效果: 尽管采用了多种优化策略,模型在原始空间的预测效果仍不理想,R²为负值表明模型预测能力有限。
UniMol预训练模型虽然能够捕获通用的分子结构信息,但可能缺乏TPA预测所需的特定电子结构特征,如:
- 共轭体系的精确描述
- 电子供体-受体相互作用
- 激发态性质相关的量子化学信息
训练集仅有615个样本,对于深度学习模型而言数据量偏小,限制了模型的学习能力和泛化性能。
当前研究仅使用Et30极性参数描述溶剂效应,未充分考虑溶剂-溶质相互作用的复杂性。
- 方法创新: 首次将UniMol预训练模型应用于TPA预测,探索了端到端学习的可能性。
- 工程实践: 通过数据预处理、模型简化等策略有效解决了实际建模中的技术问题。
- 系统比较: 全面评估了多种机器学习算法在TPA预测任务上的性能。
通过本研究,我深入了解了:
- 化学信息学中分子表示学习的原理和应用
- 深度学习模型的过拟合问题及其解决方案
- 化学数据的特殊性及相应的预处理策略
- 特征工程优化: 需要更深入理解TPA的物理化学机制,设计更有针对性的特征描述符。
- 可解释性分析: 未来将引入SHAP等工具分析特征重要性,提升模型的可解释性。
- 数据增强: 考虑通过数据增强或迁移学习策略扩充训练数据。
- 多尺度建模: 结合量子化学计算和机器学习,构建多尺度的TPA预测模型。
- 深入学习SHAP等可解释性分析方法
- 系统学习化学描述符的设计原理和计算方法
- 探索图神经网络等更适合分子数据的模型架构
- 结合领域知识设计更有效的特征融合策略
本研究虽然在预测精度上存在不足,但为利用预训练模型进行TPA预测提供了有价值的探索,为后续研究奠定了基础。通过持续学习和改进,期望在未来的工作中取得更好的成果。