

Mepylome is a high-performance Python toolkit tailored for parsing, processing, and analyzing methylation array IDAT files. Serving as a versatile library, Mepylome supports a wide range of methylation analysis tasks. It also includes an interactive GUI that enables users to generate UMAP plots and CNV plots (Copy Number Variation) directly from collections of IDAT files. Mepylome is designed for large-scale cohort processing and delivers substantial speed improvements over existing methylation analysis pipelines.
- ⚡ Very fast processing engine
- 🧬 Supports Illumina 450k, EPIC, EPICv2, 27k, MSA48, and MM285 arrays
- 📊 CNV analysis with interactive Plotly visualizations
- 🧹 Clean API for large-scale cohort processing
- 🖥️ Optional GUI for UMAP + CNV exploration
- 🧪 Built-in QC including pOOBAH detection and quality metrics
- Parsing of IDAT files
- Extraction of methylation signals
- Calculation of Copy Number Variations (CNV) with visualization using plotly.
- Interactive visualization using Plotly
- CN-summary plots
- Illumina EPIC / EPICv2
- Illumina 450k
- Illumina 27k
- Illumina MSA48
- Illumina MM285 (Mouse)
- pOOBAH-based probe calculation
- Quality metrics for samples and probes
- Heatmap of beta values of a gene or genomic region
- Methylation analysis tool with a graphical browser interface for UMAP
analysis, CNV plots and supervised classification
- Can be run from the command line with minimal setup or customized through a Python script
The mepylome documentation, including installation instructions, tutorial and API, is available at https://mepylome.readthedocs.io/
from pathlib import Path
from mepylome import CNV, MethylData
# Sample
analysis_dir = Path("/path/to/idat/directory")
sample_file = analysis_dir / "200925700125_R07C01"
# CNV neutral reference files
reference_dir = Path("/path/to/reference/directory")
# Get methylation data
sample_methyl = MethylData(file=sample_file)
reference_methyl = MethylData(file=reference_dir)
# Beta value
betas = sample_methyl.betas
# Print overview of processed data
print(sample_methyl)
# CNV anylsis
cnv = CNV.set_all(sample_methyl, reference_methyl)
# Visualize CNV in the browser
cnv.plot()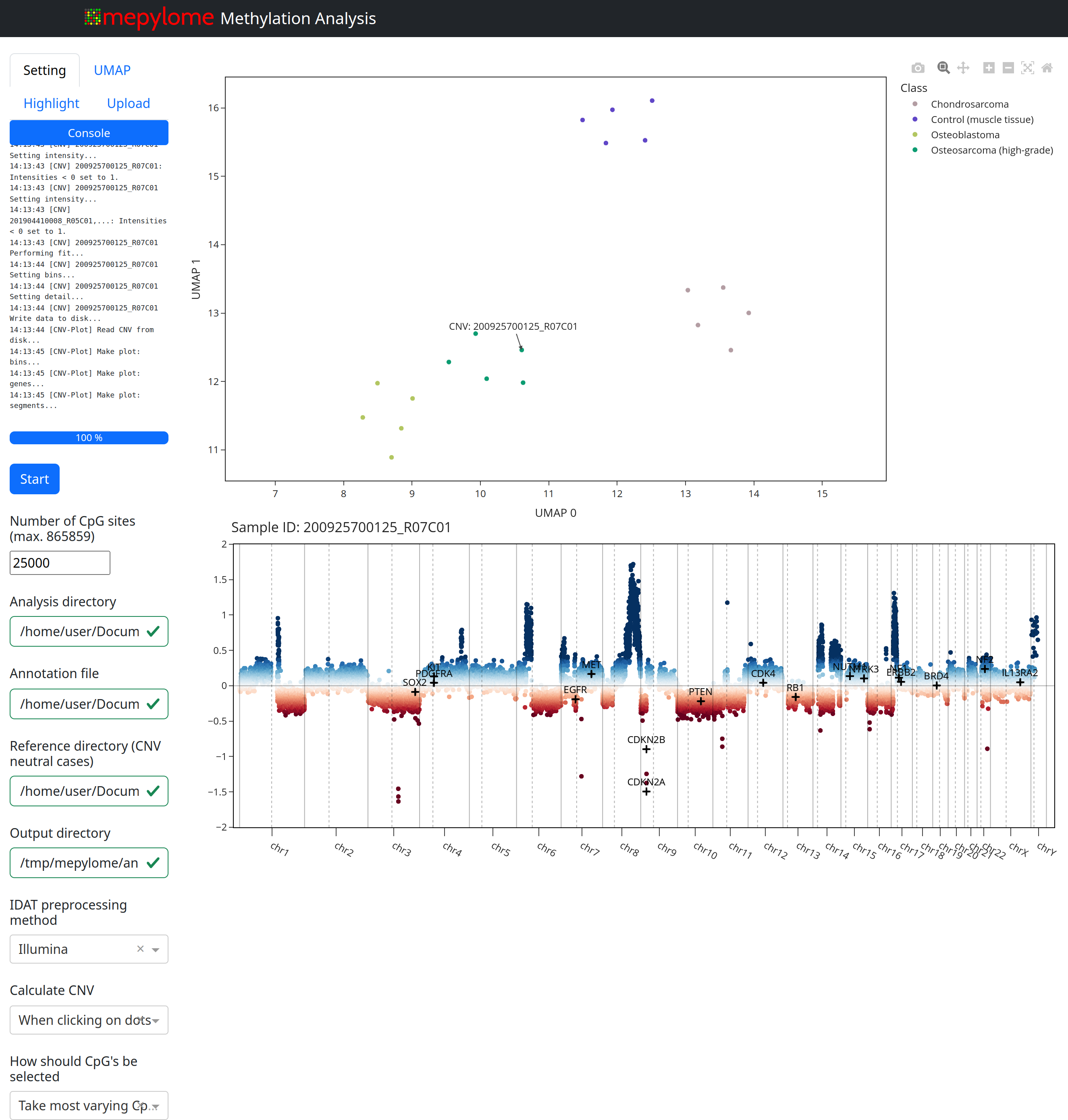
Mepylome provides a command-line interface for launching a GUI and performing methylation analysis. Ensure you have an analysis directory, a CNV reference directory, and an annotation file (located within the analysis directory). Use the following command to initiate the analysis:
mepylome --analysis_dir /path/to/idats --reference_dir /path/to/refIf you want to perform a quick test, use:
mepylome --tutorialThis command downloads sample IDAT files and provides a demonstration of the package's functionality.
You can try the tutorial directly in Google Colab-without downloading or installing anything-by clicking the link below. Please note that GUI support is limited in Colab.
See https://mepylome.readthedocs.io/ for details.
This library is described in the following peer-reviewed publication:
https://doi.org/10.1002/aisy.202500778
Contributions are welcome! If you have any bug reports, feature requests, or suggestions, please open an issue or submit a pull request.
This project is licensed under the MIT license.
Mepylome is strongly influenced by minfi and conumee2. Some functionalities, such as the manifest handler and parser, are adapted from methylprep.