![]()


A modern, performance-forward successor to samblaster for marking and removing PCR duplicates in query-grouped SAM/BAM files — streaming, BAM-native, threaded IO, and tuned to disappear into the aligner pipeline.

Visit us at Fulcrum Genomics to learn more about how we can power your bioinformatics with dupblaster and beyond.
- Coordinate-based duplicate marking, no coordinate sort required. dupblaster reads alignments directly from the aligner's output and marks duplicates in a single streaming pass over query-grouped data. This is the same approach pioneered by samblaster (Faust & Hall, Bioinformatics 2014).
- CPU-lean in the hot path. dupblaster minimizes per-record work — SIMD SAM
parsing, minimal allocations, and a hand-tuned partitioned hash table for the
coordinate index — so a
bwa-mem | dupblaster | samtools sortpipeline spends its cycles on alignment and sorting rather than on dup-marking. - Dedicated IO threads and buffers on both sides of the pipeline. Reading and
writing run on their own threads with lock-free ring buffers between them and
the worker, so dupblaster rarely blocks on IO: a brief stall in
samtools sort(e.g. flushing a sort chunk to disk) does not back-pressure the aligner. - BAM-native I/O, no SAM text adapter step. Input is auto-detected (SAM or
BAM); output is always BAM (uncompressed by default; see
--compression-level). Pair dupblaster with bwa-mem3's--bam=0flag to skip SAM-text encoding end-to-end through the aligner pipeline. - Library-aware by default, like Picard MarkDuplicates. When the header
declares more than one library (
@RG ... LB:), duplicates are called only within a library; single-library inputs are unaffected (and unchanged in speed/memory).--library-unawareforces samblaster's library-agnostic behavior. See § Library awareness. - A per-library stats TSV, ready for QC pipelines.
--stats <PATH>writes a wide TSV — one row per library — with sample, template/duplicate counts, Picard-stylefrac_duplicates, and a Lander-Waterman library-size estimate. - Modern, gnu-style CLI.
--remove-dups,--add-mate-tags,--ignore-unmated,--max-read-length,--stats, … no camelCase flags.
dupblaster is also the fastest option in the suite: on a compute-bound 8× WGS
benchmark it marks duplicates ~14× faster than Picard MarkDuplicates and
samtools markdup on x86, and ~21–25× faster on Graviton4, at a fraction of the
memory.
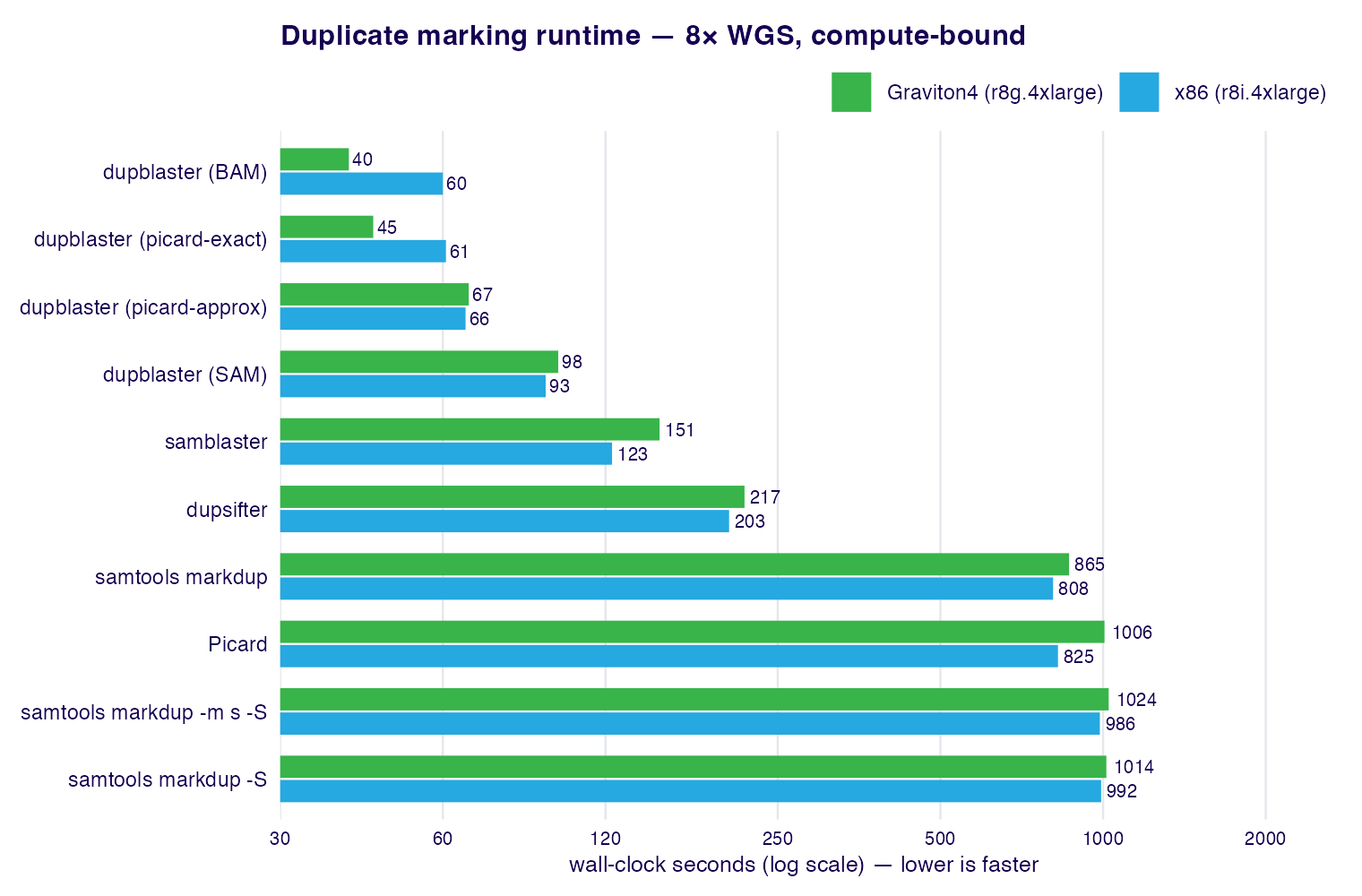
See § Benchmarks for the full per-architecture tables and methodology, and § Functional equivalence for concordance with Picard MarkDuplicates.
Jump to: Install · Quick start · Recipes · Input assumptions · CLI summary · Algorithm · Benchmarks · Limitations
# From crates.io (recommended for Rust users):
cargo install dupblaster
# Via bioconda (recommended for genomics pipelines):
conda install -c bioconda dupblaster
# From source:
git clone https://github.com/fulcrumgenomics/dupblaster.git
cd dupblaster
cargo build --release
# binary at target/release/dupblasterDrop dupblaster into the standard align → mark-dups → sort pipeline, directly after the aligner:
# With bwa-mem3 (recommended — emits uncompressed BAM with --bam=0,
# skipping the SAM-text round trip entirely):
bwa-mem3 mem --bam=0 -t 8 ref.fa r1.fq.gz r2.fq.gz \
| dupblaster --stats sample.dupblaster.tsv -o - \
| mako sort -o sample.bam -
# Or with samtools sort:
bwa-mem3 mem --bam=0 -t 8 ref.fa r1.fq.gz r2.fq.gz \
| dupblaster --stats sample.dupblaster.tsv -o - \
| samtools sort -@ 4 -o sample.bam -
# Or with classic bwa-mem (SAM output; dupblaster auto-detects):
bwa mem -t 8 ref.fa r1.fq.gz r2.fq.gz \
| dupblaster --stats sample.dupblaster.tsv -o sample.dups.bamThe pipeline above pairs dupblaster with bwa-mem31 and mako2.
All of these read query-grouped SAM/BAM and write BAM; the flags compose
freely. Multi-library inputs need no flag — dupblaster splits on @RG LB:
automatically (see § Library awareness).
# Remove duplicates instead of flagging them (leaner BAM out):
bwa-mem3 mem --bam=0 -t 8 ref.fa r1.fq.gz r2.fq.gz \
| dupblaster --remove-dups -o - \
| mako sort -o sample.bam -
# Add MC (mate CIGAR) and MQ (mate MAPQ) tags, which some downstream callers
# and UMI tools expect:
bwa-mem3 mem --bam=0 -t 8 ref.fa r1.fq.gz r2.fq.gz \
| dupblaster --add-mate-tags -o - \
| mako sort -o sample.bam -
# Exact, order-independent orphan handling (Picard's "fragments don't beat
# pairs"). Orphans are emitted at the end of the stream, so sort downstream:
bwa-mem3 mem --bam=0 -t 8 ref.fa r1.fq.gz r2.fq.gz \
| dupblaster --single-end-strategy picard-exact --tmp-dir /scratch -o - \
| mako sort -o sample.bam -
# Bisulfite / EM-seq / TAPS (directional preps): keep the two original strands
# (OT/OB) of each fragment distinct so methylation isn't lost to dup-collapsing.
# Use any bisulfite aligner that emits query-grouped output (e.g. bwa-meth):
bwa-meth.py --reference ref.fa r1.fq.gz r2.fq.gz \
| dupblaster --methylation-mode directional -o - \
| mako sort -o sample.bam -See § Methylation mode for what directional does and
why non-directional / PBAT libraries are out of scope.
Two assumptions that, if violated, either fail the run or produce wrong answers.
Every record for a given QNAME must appear in one contiguous run, with no other
QNAME's records interleaved; the order of QNAMEs relative to each other doesn't
matter. In SAM/BAM terms this is @HD SO:unsorted GO:query (equivalently
grouporder=queryname), which bwa-mem, bwa-mem3, bwa-mem2, and bowtie2
emit naturally.
dupblaster makes a best effort to catch coordinate-sorted input and fail loudly:
- Paired-end: the first QNAME's block holds only one mate-half, so dupblaster aborts with an "unmated record" error on record one.
- Chimeric or multi-mapped reads (essentially all modern WGS): a block eventually contains only secondary/supplementary alignments — its primary sits at a different coordinate — triggering a "QNAME … but no primary" error.
- Undetectable: pure single-end data with no secondary/supplementary alignments. Every block legitimately holds one primary, so the ordering is invisible and coordinate-sorted input silently produces wrong dup calls.
To dedupe a coordinate-sorted BAM, either re-sort to query-grouped
(mako sort --queryname or samtools sort -n), or use a coordinate-sort-aware
tool (samtools markdup, Picard MarkDuplicates).
dupblaster writes only BAM. The -o/--output path must be - (stdout) or end
in .bam; any other extension is rejected at startup rather than producing a
misnamed file.
Output defaults to uncompressed BGZF (level 0, "stored" blocks, same as
samtools view -u): the dominant pipeline pipes into a sort step that
recompresses anyway, so skipping the round-trip saves wall time. Use
--compression-level <0-12> when writing to durable storage or to a sink that
won't recompress.
The most common flags:
| Flag | Purpose |
|---|---|
-i, --input <PATH> |
Input SAM/BAM file (default: stdin). |
-o, --output <PATH> |
Output BAM file or - for stdout. Must end in .bam. |
-r, --remove-dups |
Drop duplicate reads from output instead of just flagging them. |
--add-mate-tags |
Add MC (mate CIGAR) and MQ (mate MAPQ) tags to all paired records. |
--ignore-unmated |
Don't abort if a primary record's mate is missing. |
--compression-level <N> |
BGZF compression level for output (0-12). Default 0 = uncompressed. |
--single-end-strategy <NAME> |
How to key single-end / orphan reads. strand-aware (default), picard-approx, picard-exact, or samblaster-legacy. See § Single-end / orphan handling. |
--methylation-mode <MODE> |
Methylation-aware keying for bisulfite / enzymatic-conversion data. Off by default. directional keeps the two original strands (OT/OB) of a fragment distinct. See § Methylation mode. |
--tmp-dir <DIR> |
Directory for the temp BAM used by --single-end-strategy picard-exact (default: $TMPDIR). |
--library-unaware |
Disable library-aware marking; use one dedup table across all reads (samblaster behavior). No effect when the header has ≤1 library. See § Library awareness. |
--stats <PATH> |
Write a per-library TSV of run-summary metrics (one row per library). |
--sample <NAME> |
Override the sample column in --stats output. |
Run dupblaster --help for the full list, including tuning knobs for
the IO ring buffers (--read-buffer-mb, --write-buffer-mb) and
BGZF CRC verification (--check-crc / --no-check-crc).
--stats <PATH> writes a TSV with one column per metric and one row per
library — in library-aware runs, one row for each @RG LB: that saw data
(the catch-all "Unknown Library" row appears only if it did); single-library
runs emit a single row. This is the format we use in our own QC pipelines —
easy to concatenate across many samples and load into pandas / data.table /
DuckDB. Give --stats a .gz (or .bgz) suffix to gzip-compress the file.
| Column | Meaning |
|---|---|
sample |
From --sample, or comma-joined @RG SM: values, or empty. |
library |
Library name (@RG LB:), Unknown Library, or All Reads under --library-unaware. |
dupblaster_version |
Version of dupblaster that produced this row. |
total_templates |
Templates (QNAMEs) seen with a usable primary. |
duplicate_templates |
Templates marked as duplicates of another template. |
frac_duplicates |
Picard-style read-level fraction: (orphan_dups + 2*pair_dups) / (orphan_reads + 2*pair_reads). |
mapped_pairs |
Templates with both reads mapped. |
duplicate_pairs |
Mapped pairs marked duplicate. |
mapped_orphans |
Templates with exactly one read mapped. |
duplicate_orphans |
Mapped orphans marked duplicate. |
unmapped_orphans |
Templates with one read present and unmapped (no mapped mate). |
unmapped_pairs |
Templates with both reads unmapped. |
unmated_templates |
Templates with a stray half (skipped unless --ignore-unmated). |
estimated_library_size |
Lander-Waterman estimate of unique molecules; empty when not estimable. |
dupblaster uses the same coordinate-based dedup approach as samblaster (see Faust & Hall, Bioinformatics 2014 for the full algorithm): for each template, compute a key from the 5'-aligned positions and strands of the primary first-of-pair and second-of-pair (or the unpaired primary), and call a template a duplicate if its key has been seen before. Secondary and supplementary alignments inherit the duplicate flag from their primary.
The coordinate index is a partitioned hash table sized to the genome's contig list. The per-contig position cap is 2^31 − 1 bp (~2.15 Gb) — this is the SAM/BAM format's own limit, not a dupblaster constraint. Every realistic reference genome fits well under that: human chr1 is 0.25 Gb, the largest plant chromosomes (wheat) are under 1 Gb, axolotl tops out at around 3 Gb chromosomes (which would hit the cap — file an issue if this affects you). The number of contigs is unbounded.
Comparisons account for soft-clipping at the 5' end, so two reads with different clipping but the same true alignment start are correctly identified as duplicates.
--single-end-strategy selects how single-end reads and orphans (a
mapped read whose mate is unmapped) are keyed in the dedup table.
Four strategies are supported:
strand-aware(default). The dedup key is the strand-specific 5'-aligned position. A forward orphan and a reverse orphan at the same 5' coord are not duplicates. This matches Picard MarkDuplicates'fragSortkeying at the single-end level and is the right answer for short-read PE data.picard-approx. Strand-aware key plus a Picard-style cross- check: each end of every fully-mapped PE pair is also registered in a fragment-level table, so a later orphan / single-end read at the same 5' coord is marked as a duplicate of the pair. This approximates Picard's "fragments don't beat pairs" rule in a single streaming pass — approximate because an orphan that arrives before its corresponding pair passes through as non-dup. Roughly doubles dupblaster's memory footprint at run time. Recommended when you want Picard-equivalent dup partitions and have the memory headroom.picard-exact. The same "fragments don't beat pairs" rule aspicard-approx, but exact and order-independent. dupblaster runs two passes: fully-mapped and unmapped pairs stream straight to the output, while every mapped-orphan / single-end read is buffered to a temporary uncompressed BAM (see--tmp-dir). After the pair pass, the pair table is consumed into a fragment table holding the 5' position of every paired read end, and the buffered fragments are re-read and marked against it — so an orphan is marked a duplicate of a pair regardless of which came first in the stream, unlikepicard-approx. Two trade-offs: (1) buffered fragments are emitted at the end of the output stream rather than in input order (re-sort downstream if order matters — most pipelines already do), and (2) it writes a temporary on-disk copy of the fragment reads. Intended for paired data, where orphans are a small fraction; on single-end-only libraries it would buffer the entire input. Matches Picard's fragment dup counts / partitions exactly; it does not attempt to reproduce Picard's choice of which read in a duplicate set is the representative.samblaster-legacy. samblaster's post-v0.1.25 (Feb 2020) behavior: leftmost-aligned reference coordinate with the strand bit dropped, so a forward orphan and a reverse orphan at the same leftmost-aligned position collide. Produces false positives on short-read PE data (two distinct molecules sharing only their leftmost-aligned coord on opposite strands are marked dup). Provided for byte-compatibility with samblaster output on long- read singleton workflows where it was originally validated; not recommended otherwise.
--methylation-mode directional adapts duplicate marking for bisulfite
and enzymatic-conversion libraries (WGBS, EM-seq, TAPS). In these data
the two strands of a fragment — the original-top (OT/CTOT) and
original-bottom (OB/CTOB) — carry independent methylation and must be
counted as separate molecules, not duplicates of each other.
Standard WGS keying canonicalizes each pair to a coordinate-ordered signature (leftmost end first). At a given locus the OT and OB fragments occupy the same two coordinates, so canonicalization gives them the same key and the second is wrongly marked a duplicate. Directional mode keys the pair in template order instead — first-of-pair into slot A, second-of-pair into slot B, with no coordinate swap. Because directional preps ligate adapters to the intact double-stranded fragment before conversion, first-of-pair is locked to a consistent end of the original strand, so:
- the OT and OB pairs produce different keys (their first-of-pair reads sit on opposite ends and strands) and are kept distinct, while
- genuine PCR copies of one strand reproduce the same template-order geometry and still collapse.
This holds across all pair orientations (FR/RF/FF/RR) and cross-contig chimeras: orientation is simply part of the key. The single-end / orphan path is unchanged — it is already strand-aware, so OT/OB orphans stay separate in every mode.
Scope. Only directional libraries are supported. Non-directional
/ PBAT libraries (where the first-of-pair-to-strand relationship is
not fixed) are out of scope: correct keying there needs per-read strand
tags plus a canonicalize-within-strand key, which is a separate piece
of work. --methylation-mode pbat is intentionally rejected rather than
silently mis-handled. The flag is also independent of (and composes with)
--single-end-strategy, --remove-dups, and --add-mate-tags.
Duplicates are a property of a library (a PCR-amplified pool), not of the genome: two reads at the same coordinates from different libraries are independent observations, not copies of one molecule. Picard MarkDuplicates keys on the library; samblaster ignores it. dupblaster follows Picard by default.
Library membership comes from each read's RG:Z tag, mapped through the
header's @RG ... LB: field. Read groups that share one LB are one library;
reads with no RG, an RG absent from the header, or an @RG line with no
LB share a single "Unknown Library" bucket. The dedup state is then
partitioned into one independent table per library.
This activates only when the header declares more than one distinct LB.
With zero or one library, there's nothing to separate, so dupblaster runs in
single-table mode — identical results, and identical speed and memory, to
before (no per-read RG scan). Pass --library-unaware to force single-table
mode even with a multi-library header (samblaster's behavior).
Memory. Each library's table is allocated lazily (only libraries that
actually appear cost anything), and per-cell pre-sizing is scaled down by
ceil(√library_count). The empty-table baseline therefore grows roughly with
the square root of the library count rather than linearly, and the stored
signatures — the part that scales with data — are essentially conserved when a
fixed amount of sequencing is split across libraries (identical fragments
rarely recur across independent preps). In practice a 3-library run and a
1-library run over the same data use within a few percent of the same memory.
Benchmarked on two AWS instance classes — r8i.4xlarge (Intel x86) and
r8g.4xlarge (Graviton4), each 16 vCPU / 123 GiB RAM. The reproducible
Snakemake pipeline in
benchmark-pipeline/ drives the runs and
bench-compare/ evaluates functional equivalence against
Picard MarkDuplicates. The input is 8× WGS downsampled from NYGC 1000 Genomes
sample HG03953 (bwa-mem aligned, retaining supplementary alignments): a 67 GB
query-grouped BAM, or 83 GB as SAM text.
The streaming tools here — dupblaster and samblaster — process uncompressed data faster than all but the fastest local SSDs can supply or absorb it. They are built for a different deployment: a single pipe between an aligner and a sorter, where the surrounding stages keep the stream moving and storage is never the bottleneck. Benchmarking them against a disk would measure the disk, not the tool, and would not reflect how they are actually run.
We model that no-I/O-bottleneck case directly. Each tool's input is pre-warmed
into the page cache so the timed read runs at RAM speed, and each tool writes its
output to a FIFO drained to /dev/null (the writer thread still serializes every
record, so its CPU is counted). Format conversion and any sort a tool requires
happen outside the timed window.
The numbers below are dupblaster 0.1.0, a single replicate, taken 2026-06-13; the
raw collated reports are committed in
benchmark-pipeline/published-results/.
x86 — r8i.4xlarge (Intel, 16 vCPU)
| Tool | Runtime (s) | CPU (s) | RSS (MB) | × fastest |
|---|---|---|---|---|
| dupblaster 0.1.0 (BAM) | 60.0 | 68.6 | 1248 | 1.0× |
| dupblaster 0.1.0 (picard-approx) | 66.1 | 91.0 | 2314 | 1.1× |
| dupblaster 0.1.0 (picard-exact) | 60.8 | 71.2 | 1466 | 1.0× |
| dupblaster 0.1.0 (SAM) | 93.0 | 125.3 | 1229 | 1.5× |
| samblaster 0.1.26 | 123.4 | 123.4 | 1425 | 2.1× |
| dupsifter 1.3.0 | 203.2 | 203.1 | 3114 | 3.4× |
| Picard 3.4.0 | 825.2 | 1250.1 | 8578 | 13.8× |
| samtools markdup 1.23 | 808.2 | 795.2 | 218 | 13.5× |
samtools markdup 1.23 (-S) |
991.9 | 948.1 | 228 | 16.5× |
samtools markdup 1.23 (-m s -S) |
986.5 | 955.6 | 228 | 16.5× |
Graviton4 — r8g.4xlarge (Arm, 16 vCPU)
| Tool | Runtime (s) | CPU (s) | RSS (MB) | × fastest |
|---|---|---|---|---|
| dupblaster 0.1.0 (BAM) | 40.2 | 60.0 | 1236 | 1.0× |
| dupblaster 0.1.0 (picard-approx) | 67.0 | 96.1 | 2327 | 1.7× |
| dupblaster 0.1.0 (picard-exact) | 44.6 | 62.9 | 1438 | 1.1× |
| dupblaster 0.1.0 (SAM) | 98.1 | 115.0 | 1228 | 2.4× |
| samblaster 0.1.26 | 151.1 | 151.1 | 1424 | 3.8× |
| dupsifter 1.3.0 | 217.0 | 216.9 | 3113 | 5.4× |
| Picard 3.4.0 | 1006.1 | 1504.1 | 8523 | 25.0× |
| samtools markdup 1.23 | 865.2 | 864.0 | 217 | 21.5× |
samtools markdup 1.23 (-S) |
1014.2 | 1012.8 | 227 | 25.2× |
samtools markdup 1.23 (-m s -S) |
1024.3 | 1017.7 | 227 | 25.5× |
samtools markdup's timed window excludes the fixmate -m + coordinate-sort
prep it requires; the other tools consume the query-grouped input directly.
dupblaster is ~14× faster than Picard and samtools markdup on x86 and ~21–25× faster on Graviton4. The margin is larger on Graviton4: dupblaster's lighter per-record work scales across the cores (40 s vs 60 s on x86), while Picard's heavier per-read cost does not (1006 s vs 825 s). Resident memory differs similarly — dupblaster holds ~1.2 GB versus Picard's ~8.5 GB.
dupblaster exposes two orthogonal options. Input format affects only speed: BAM
is the native on-disk shape, while the SAM path must text-parse the stream
(~1.5–2.4× the wall time, identical marking). Single-end strategy affects only
orphan handling (see § Single-end / orphan handling):
the default strand-aware is fastest,
picard-approx adds a streaming pair-end cross-check, and picard-exact
buffers orphans for an order-independent second pass. All three mark paired
reads identically; pick a strategy for orphan fidelity, a format for speed.
| Tool | PE concordance | SE/orphan concordance | Supp. marked |
|---|---|---|---|
| Picard 3.4.0 | reference | reference | only on query-grouped input |
| dupblaster 0.1.0 (strand-aware) | 100% | 75.5% | yes |
| dupblaster 0.1.0 (picard-approx) | 100% | 95.9% | yes |
| dupblaster 0.1.0 (picard-exact) | 100% | 100% | yes |
| samblaster 0.1.26 | 100% | 75.6% | yes |
| dupsifter 1.3.0 | 86.9% | 75.5% | yes |
| samtools markdup 1.23 | 99.9% | 99.8% | no |
samtools markdup 1.23 (-S) |
99.9% | 99.8% | yes |
samtools markdup 1.23 (-m s -S) |
100% | 99.8% | yes |
Concordance is set-equivalence, not per-read agreement. We tag every Picard primary with a canonical fragment key (unclipped 5′ position + strand), so a duplicate set is all templates sharing a key — a pair keys on the sorted keys of both ends, an orphan on its one mapped end. A tool is concordant on a set when it marks the same number of templates in that set as duplicates as Picard does: same group, same count, regardless of which template each tool elects as the representative. PE counts both-ends-mapped sets; SE/orphan counts one-end-mapped sets. "Supp. marked" is whether the duplicate flag propagates from a primary to its supplementary alignments (the benchmark data has supplementary but no secondary alignments).
-
samblaster / dupblaster (strand-aware) — identical to Picard on every paired set. The orphan gap is one effect: Picard additionally cross-checks an orphan against the 5′ positions of mapped pairs and marks it a duplicate of a pair; the strand-aware key does not. dupblaster's
picard-approxandpicard-exactstrategies add that cross-check (approx in one streaming pass; exact order-independently), raising orphan concordance to 95.9% / 100%. -
dupsifter — paired concordance is only 86.9% because its signature is strand-of-origin-aware (inherited from its WGBS design) and does not collapse FR/RF orientation. For a fragment captured from both strands at the same coordinates, Picard counts one duplicate set, but dupsifter keeps one representative per orientation — marking exactly one fewer duplicate in every affected set.
-W(WGS mode) disables only the bisulfite-strand inference, not this orientation split, so the gap persists on plain WGS. -
samtools markdup — paired concordance is 99.9% in its default
-m t(template) mode; the few disagreements are FF/RR (same-strand) and inter-chromosomal pair geometries, where template mode folds R1/R2 (first/second-in-template) identity into the key and so diverges from Picard. Running-m s(sequence mode) keys on the unclipped 5′ ends without that distinction — coordinate-canonical like Picard and dupblaster — and closes the gap to 100% paired concordance (the-m s -Srow), at no meaningful runtime cost. It cross-checks orphans like Picard (99.8%). By default it does not mark the secondary/supplementary alignments of a duplicate;-Senables that, at a runtime cost. This matters for short-read structural-variant callers, which read supplementary alignments as breakpoint evidence: when the supplementary records of a dup-marked template go unflagged, they count as independent observations of what is really one PCR-amplified event, inflating breakpoint support. dupblaster (and samblaster) propagate the primary's flag to supplementary alignments by default, avoiding this; withsamtools markduppass-S(or re-flag downstream).
- Streaming, non-UMI data — use dupblaster. It is the fastest option and
reads directly from the aligner with no coordinate sort. Paired marking is
identical to Picard, and
picard-exactalso matches Picard on orphans. - Input not query-grouped — use Picard. It coordinate-sorts internally and adds optical-duplicate detection and per-read duplicate-set tags.
- Coordinate-sorted input that must stay a streaming pass — use
samtools markdup. It marks duplicates in a single pass
over coordinate-sorted BAM without re-grouping. Note its FF/RR and
inter-chromosomal differences from Picard, and pass
-Sfor secondary/supplementary marking. - UMI libraries — use fgumi. It distinguishes true PCR duplicates from coincidental position collisions; positional dedup cannot.
- No optical / sequencing duplicate detection.
- No UMI awareness — use fgumi for UMI-aware dedup.
- Methylation mode (
--methylation-mode) supports directional libraries only (WGBS / EM-seq / TAPS); non-directional / PBAT is not supported.
For users coming from samblaster, the high-level changes are:
- Modern CLI: GNU-style
--kebab-caseflags (e.g.--remove-dups, not--removeDups). - BAM-native: input is SAM or BAM; output is always uncompressed
BAM. No
samtools viewadapter step. - No SV-extraction flags (
-d,-s,-u,-a,-e): dropped. - Library-aware: duplicates are called within a library (
@RG LB:) by default, like Picard; samblaster is library-agnostic.--library-unawarerestores the samblaster behavior. - Larger genomes: no hardcoded position cap.
- Threaded IO: dedicated read and write threads with ring buffers, so the worker doesn't block on pipe stalls.
- Per-library stats TSV: structured
--statsoutput for QC pipelines, one row per library. - No header preservation guarantee:
@PGrecords are auto-chained viaPP:(samblaster does not chain). - Idempotent dup flag: dupblaster overwrites
FLAG_DUPLICATEon every output record with the current run's decision (matching Picard and samtools markdup); samblaster ORs it in, preserving prior markings. Overwrite is idempotent across re-runs (bwa mem | dupblasterandbwa mem | dupblaster | dupblasterproduce identical output); OR is not.
See CONTRIBUTING.md for development setup, the test suite, and release flow.
MIT. See LICENSE.
dupblaster is inspired by, and adapts the coordinate-based duplicate detection algorithm of, samblaster by Greg Faust and Ira Hall. If you use dupblaster in published work, please cite the samblaster paper:
Faust, G.G. and Hall, I.M., SAMBLASTER: fast duplicate marking and structural variant read extraction, Bioinformatics 30(17): 2503-2505 (2014). doi:10.1093/bioinformatics/btu314