During my time at IPK, Germany (June & July 2025), we sequenced and assembled a barley landrace following the TRITEX protocol.
This repository contains the scripts used to assemble the genome from HiFi and Hi-C reads.
The TRITEX protocol is well-documented and clearly explained. However, executing the code is not always straightforward. As of now, I understand that the Genetics and Genomic Crop Research group is actively working to improve and update the protocol, making it easier to execute.
In the meantime, this repository provides the scripts and Conda environments I used throughout the entire process, which may help others attempting a similar assembly.
Contact me for any doubt: [email protected] or find me on twitter
We will refer to our barley landrace as GDB_136, used MorexV3 as reference genome, and we based our work on the original TRITEX repository. However, we used modified scripts and Bash code to better follow and adapt the pipeline.
Clone the original repository and update the scripts according to your software paths, project names, data specifications, and locations.
Below is the file and directory structure of the working environment:
tritex_barley_assembly/
├── 01.5_splitFASTQ.sh
├── 01_bam2fastq.sh
├── 02_contig_assembly.sh
├── 03_check_contig_stats.sh
├── 04_gfa2fa.sh
├── 05_digest_enzyme_HiC.sh
├── 06_HiC_mapping.sh
├── 07_prepare_reference.sh
├── 08_guide_map_create.R
├── 09_map_guide_map.sh
├── 10_create_assembly_object.R
├── 11_break_chimeric_scaffold.R
├── 12_fix_chimeras_scaffold.R
├── 13_make_HiC_matrix.R
├── 14_second_round_scaffold.R
├── 15_500kb_scaffolds.R
├── 16_collinear_plot.R
├── 17.05_MapInspectorShiny.R # This steps are optional, and up to today, I have not been able to run it successully
├── 17_MapInspectorShiny.R # This steps are optional, and up to today, I have not been able to run it successully
├── 18_pseudomol_stats.R
├── 19_compile_pseudomol.R
#
# # Until here you can see our own scripts to follow the pipeline. Down here find all data, outputs and TRITEX repository.
#
├── assembly/ # Empty folder to store assembly data
├── HiC/ # Folder containing the raw HiC data
├── Hi-Fi/ # Folder containing the raw Hi-Fi data
├── HOWTO.txt
├── mapping/ # Empty folder that will contain the HiC mapped data to the scaffolds
├── MorexV3.fa # The reference genome of the species
├── output/ # Empty folder to store output files
├── pseudomolecules/ # Empty folder to store pseudomolecules compiled
├── test_env.yml # yml file to create a conda environment to perform (almost) whole pipeline
└── tritexassembly.bitbucket.io/ # The repository with the scripts, download and modified from IPK repositoy
Feel free to adapt this structure to your own naming conventions or computational setup. The initial data folders should look like:
IPK/
├── HiC/
│ ├── 3401768_GDB136_S3_L002_R1_001.fastq.gz
│ └── 3401768_GDB136_S3_L002_R2_001.fastq.gz
└── Hi-Fi/
└── Gm84096_250523_032626_s1.hifi_reads.bam
Note: you may have the Hi-Fi data in fastq format. This is allright too
Download as well the reference genome in the main folder, MorexV3 in this case for barley.
Start cloning the repository:
git clone https://github.com/jsarriaa/TRITEX_Barley_assembly_protocol.git
And create and activate the environment to work on:
conda env create -f test_env.yml
conda activate IPK-tritex
NOTE
TRITEX was originally prepared to work with some proprietary software such as NovoSort. In this case, we have adapted it to work with open-source alternatives.
Initially, the pipeline was designed for macOS systems, with some scripts using macOS-specific syntax (e.g.,
.zshinstead of.sh). Some of those scripts have been modified to work on Linux servers.R scripts were originally intended to be run in RStudio. However, this may not be convenient when working on remote servers (via SSH), where graphical interfaces are unavailable. All custom R scripts have been adapted to run in the command line using
Rscript. Feel free to adapt them back to RStudio if you prefer working in that environment.
NOTE 2
These are the recommended software versions for the TRITEX pipeline, as suggested by the authors:Tools
- Z shell, v5.0.2
- GNU Parallel, v20150222
- Hifiasm, v0.15.1-r334
- gfatools, v0.5
- SAMtools, v1.9
- bgzip & tabix (via BCFtools), v1.9
- Seqtk, v1.0-r76
- Bedtools, v2.26.0
- R, v3.5.1
- Pigz (parallel gzip), v2.3.4
- Minimap2, v2.14
- Cutadapt, v1.15
- EMBOSS, v6.6.0
- BBMap, v37.93
- faToTwoBit & twoBitInfo (UCSC tools), v3.69
If you don't have access to the commercial NovoSort, you can follow the alternative SAMtools-based approach provided in the TRITEX shell scripts.
Required R Libraries
data.table, v1.11.8stringi, v1.2.4igraph, v1.2.2zoo, v1.8-3parallel(included in R-core ≥ 2.14)
You may have the Hi-Fi data in a .bam file instead of a compressed .fastq. Run this script to addapt the data for TRITEX protocol:
## Edit variables on the script
./01_bam2fastq.sh
Hi-Fi is heavy, and split it make the scaffold building easier and more optimal. Run:
./01.5_splitFASTQ.sh Hi-Fi/<input.fastq>
It is prepaired to split the file into 20 parts. Edit line parts=20 if you desire.
Using Hifiasm build contigs from the Hi-Fi.
Set up properly the directory and variables and run:
./02_contig_assembly.sh
Check and generate your contigs stats, runnning:
./03_check_contig_stats.sh
It will outcome files summarizing the generated contigs from hifiasm, and the uncontigs ones as well.
Interesting data, as the N50, N90, mean and contig size can be extracted from it.
Then, transform de gfa file into fasta format, also seting contigs names:
./04_gfa2fa.sh
First, you need to digest in silico the hifiasm assembly with the restriction enzyme used for the preparation of the Hi-C libraries.
At the following script remember to properly indicate which enzyme has been used to prepare your library.
./05_digest_enzyme_HiC.sh
Take care about the script at
bitbucket/shell/digest_emboss.zsh. You may have to change the paths for your exectuables, such as restrict, bedtools and rebase.
Now, map the HiC data. Ensure at the script set up the variables, enzyme used, and executables paths. Output will be stored at mapping folder.
./06_HiC_mapping.sh
From a reference genome, create a guide map table. Take a look with calm to the TRITEX guide map documentation to understand the database.
Obtain from a reference genome a single copy 100bp regions. In this case, barley, we are using MorexV3.
bitbucket/miscellaneous/mask_assembly.zsh script requires to change the paths to the executables.
Also remember to change at 07_prepare_reference.sh your own paths and variables and run:
./07_prepare_reference.sh
After masking, prepare the guide map. Take a look and set up the variables, and prepare the output files names.
./08_guide_map_create.R
Modify with your variables and run:
./09_map_guide_map.sh
From now on, work will be mainly performed at R. You must start creating the assembly object as an R database (remember to set variables before running):
Rscript 10_create_assembly_object.R
All ouptuts from now on will be located at /pseudomolecules folder. Future steps will generate more databases. It is recomended to name databases to be able to distingish (for instance, adding the date).
By mistake scaffolds wrongly constructed may appear (in example, sequences from diference chromosomes but a similar region might be combined together, and must be corrected manually).
For this, generate diagnostic plots:
Rscript 11_break_chimeric_scaffold.R
It generate plots for all contigs of more than 1 Mb (do not forget to change variables).
The curation step must be performed by you manually. Take a carefully view to the generated pdf. You should find chimeras such as pages 3, 6 and 9 of this example.
Then, specify the bin positions in where the chimeras have to be broken. Do it editing the script 12_fix_chimeras_scaffold.R. Modify the variable i as the page you found an chimera, and and set and estimated range where you observe the chimera. Add as many times you need this block of code. Once you have all ready, run:
Rscript 12_fix_chimeras_scaffold.R
Now, you will have a new assembly object. Check at the new pdf produced that now the chimeric scaffolds are now properly divided, and carry on:
Prepare the variables at the file and run the script:
Rscript 13_make_HiC_matrix.R
Check the produced pdfs and excel sheet.
If everything is allright, carry on. Let's do a second round of looking to chimeras. Now, decreasing down to at least 500kb scaffolds. Prepare and run the script to first fixing chimeras, or plotting specific scaffolds:
Rscript 14_second_round_scaffold.R
Then: (remember to use the new updated database, and set up variables)
Rscript 15_500kb_scaffolds.R
This step is not mandatory but recomended. Looks for collinearities between the scaffolds, helping to spot inverted or missplaced things. Pay attention at the number of chromosomes that your species have. Once set everything up, run:
Rscript 16_collinear_plot.R
IMPORTANT NOTE: Those following scripts have not been tested. Use on your own knowledge. Recomended trying to follow the original TRITEX guide.
At the colinear plots you can observe inverted scaffolds, like at this example.
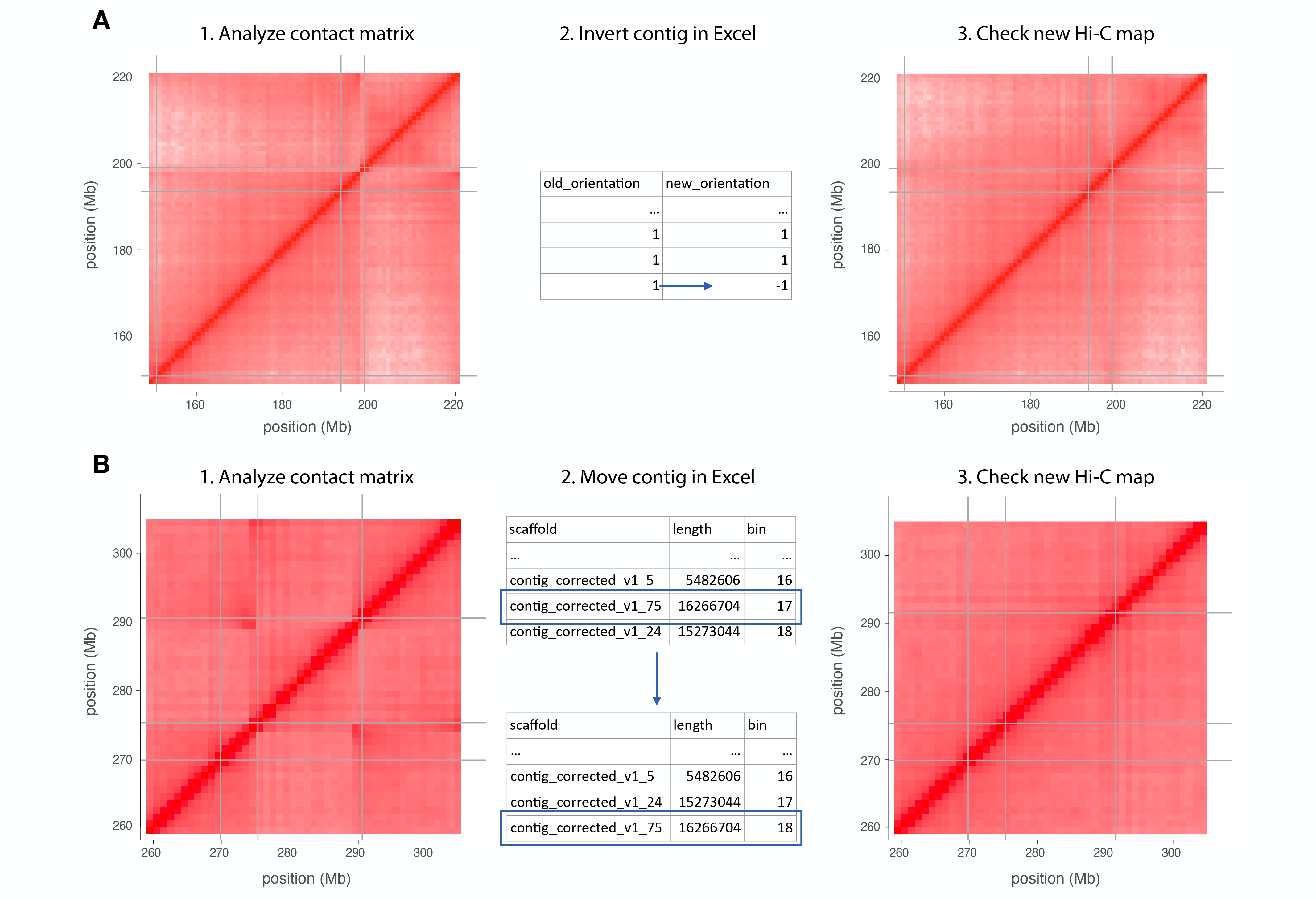{kind=link}
When you spot one of this scaffolds to be corrected, go to the assembly excel file (produced at steps as the 15 script) and modify the new orientation value for the scaffold.
Download and use Shiny, you can follow this manual recomended by TRITEX pipeline.
You can try to run 17.05_MapInspectorShiny.R and 17_MapInspectorShiny.R with Rscript but note I have not tested them properly, because Map Inspector Shiny can not be installed with the specifications provided by the pipeline documentation. Try it on your own if you consider.
Note: at scripts I indicate you should work with the corrected database by Map shiny inspector if you have, if not (as me) work with the obtained after the script nº15.
First, lets get some data from the outcome pseudomolecules. Prepare the script with the variables (look over the script that you have to specify the number of chr of your species):
Rscript 18_pseudomol_stats.R
And compile it:
Same, set uo variables and work over the corrected database You must prperly modify the file:
/tritexassembly.bitbucket.io/R/pseudomolecule_construction.Rand edit thecompile_psmolfunction with the proper paths as variables.
Rscript 19_compile_pseudomol.R
NOTE: take a lot of care about the executable version of samtools. It is different (and must be) than last versions and the recomended one. It worked for me installing samtools version 1.21 in another path different and used it at this step, while the rest of the pipeline only worked for me at version 1.22.