After almost two years since the last stable release of anvi'o, we are happy to announce anvi'o v9 with the code name, "eunice".
After about 2,400 changes in the anvi'o repository that introduced over 25,000 new lines of code to the platform, this stable release of anvi'o represents significant advancements over v8, and introduces many new programs, new features, performance improvements, and fixes for known bugs.
The following document intends to give you a summary of the notable changes that come with eunice.

The code name honors Eunice Newton Foote, an American researcher who discovered the link between atmospheric carbon dioxide concentrations and Earth's temperature. This groundbreaking discovery emerged from a simple yet powerful experiment: Foote filled one glass cylinder with carbon dioxide and another with ordinary air, then measured their temperatures after exposing them to sunlight. She observed that the cylinder containing carbon dioxide trapped much more heat and remained hot much longer than the control. From this simple observation, she reached an elegant conclusion that established the first connection between carbon dioxide gas and planetary warming. Despite the profound implications of her work, Foote was not able to communicate her findings to the members of the American Association for the Advancement of Science (Joseph Henry presented Foote's findings to the members of AAAS instead), nor was she recognized as the founder of climate science (that credit went to John Tyndall instead). Her erasure from history was largely due to women's exclusion from the scientific establishment of her era. We were able to correct the record when retired petroleum geologist Raymond P. Sorenson rediscovered her work after 150 years and sparked renewed recognition of her pioneering role in climate science by publishing an article in 2011 titled "Eunice Foote's Pioneering Research On CO2 And Climate Warming". A correction in this instance was only possible thanks to the records of Foote's work that happened to survive, which is a sobering reminder that for every erased contribution we can recover, many others were likely lost forever. As a generation that experiences the implications of the increasing temperatures her work predicted first-hand, we join others who finally recognize Eunice Newton Foote for her foundational contribution to our understanding of climate change.
The code name was a suggestion by Dr. Sofia Ibarraran-Viniegra, managing director of the NSF Science and Technology Center, Chemical Currencies of a Microbial Planet (C-CoMP). The release notes were written by Meren, Iva Veseli, Florian Trigodet, and Samuel Miller, who are among the developers of anvi'o. For a complete list of anvi'o developers and contributors, see https://anvio.org/people/.
A quick overview of the new anvi'o programs and artifacts in eunice
The new version of anvi'o comes with a bunch of new programs, increasing the total number of all programs in the anvi'o ecosystem to 176:
- anvi-compute-rarefaction-curves by Meren (@meren) and Alex Henoch (@ahenoch) that computes rarefaction curves and Heaps' Law fit for a given pangenome.
- anvi-display-codon-frequencies by Meren and Samuel Miller (@semiller10) that displays codon frequency statistics across genes in a given genome in the anvi'o interactive interface.
- anvi-draw-kegg-pathways by Samuel Miller that visualizes user metabolism data in the context of KEGG pathways to show the coverage of the enzyme pool.
- anvi-export-gene-clusters by Meren that exports gene clusters in a pan-db as a three-column, TAB-delimited file that associates each gene call in each genome with a gene cluster.
- anvi-import-metabolite-profile by Samuel Miller that imports metabolite abundance data into a profile database for downstream analyses.
- anvi-import-protein-profile by Samuel Miller that imports protein abundance data from (meta)proteomics surveys into a profile database.
- anvi-predict-metabolic-exchanges by Iva Veseli (@ivagljiva) that predicts which metabolic compounds are potentially exchanged between organisms via pairwise reaction network comparisons.
- anvi-reorient-genomes by Meren and Alex Henoch that implements a comprehensive solution to re-orient and/or scaffold genomes or metagenome-assembled genomes against a de-novo identified or user defined reference genomes.
- anvi-script-find-misassemblies by Florian Trigodet (@FlorianTrigodet) that reports errors in long-read assembly outputs using read-recruitment information.
- anvi-script-gen-defense-finder-models-to-hmm-directory by Georges Kanaan (@Ge0rges) that generates an hmm-source artifact from the MDMParis Defense Finder Models that can be used by anvi-run-hmms.
- anvi-script-hmm-to-hmm-directory by Georges Kanaan that generates an hmm-source artifact from any given single HMM model that can be used by
anvi-run-hmms. - anvi-script-reformat-bam by Andrea Telatin (@telatin) that reformats a BAM file to match the updated sequence names after running anvi-script-reformat-fasta.
And a few new artifacts, increasing the total number of all artifacts in the anvi'o ecosystem to 141:
- contig-rename-report-txt
- gene-clusters
- gene-clusters-txt
- hmm-file
- kegg-pathway-map
- kgml-walker
- pfam-accession
- rarefaction-curves
- equivalent-compounds-txt
- genome-pairs
- metabolite-exchange-predictions
In addition, this release makes significant updates to,
- anvi-run-workflow, which now has new workflows for ecophylo and sra_download by Matt Schechter, and the metagenomics workflow can now work with long reads (#2479 by Florian Trigodet; more details below).
- anvi-run-cazymes now works with version 13 of the CAZymes database (#2460 by Matt Schechter).
- anvi-run-ncbi-cogs now works with COG24 (#2419 by Meren), archaeal COGs (#2136 by Iva Veseli), and can accept FASTA files of amino acid sequences as input (#2155 by Georges Kanaan).
- anvi-estimate-scg-taxonomy can now work with multiple genomes (#2249 by Iva Veseli).
- anvi-get-sequences-for-gene-calls can now report all functions for all genes (#2424 by Benjamin Luke Coltman (@bcoltman)).
- Anvi'o can now better estimate the expected number of bacterial/archaeal genomes in a metagenomic assembly (#2235 by Iva Veseli).
With new contributors,
- Andrea Telatin made their first contribution in #2165
- @afuetterer made their first contribution in #2191
- Oleg Vlasovets (@Vlasovets) made their first contribution in #2236
- Sebastian Cristian Treitli (@treitlis) made their first contribution in #2276
- @bernhardkaindl made their first contribution in #2326
- Benjamin Luke Coltman made their first contribution in #2336
- Dylan Mankel (@mankeldy) made their first contribution in #2431
Finally, we thank Dylan Mankel, @afuetterer](http://anvio.org/people/afuetterer, Georges Kanaan, and @bernhardkaindl for improving our documentation at https://anvio.org and/or in the anvi'o codebase, and for all important bug fixes and sanity checks by Georges Kanaan (#2175), Sebastian Cristian Treitli (#2276), and Moritz Buck (@moritzbuck) (#2442).
You can see the v9 full changelog compared to v8 via v8...v9. The following sections in this document will highlight some of the most critical changes in anvi'o since the last release.
Updates to the metabolism estimation framework
-
KEGG module estimates from the interactive interface. The codebase underlying the likes of anvi-estimate-metabolism has been modularized to allow for better programmatic access to estimates of KEGG module completeness and copy number (#2477 by Meren). The result? Now you can have on-the-fly module completeness estimates for your bins in the anvi'o interactive interface, whether your bins describe contigs in a genome or assembly, or gene clusters in a pangenome.
Watch the following YouTube video to see it in action in pangenomics:
-
Expanded KEGG KOfam models with 'no-threshold KOs'. The metabolism suite of programs has a new option,
--include-nt-KOsby Iva Veseli, that allows one to annotate and utilize KEGG Orthologs that are included in the KEGG GENES database but that do not have a profile HMM with a bit score threshold provided by KEGG. We call these 'no-threshold KOs' or 'nt-KOs', and using the new flag may result in a small increase in KO annotations or KEGG module completeness estimates.When you use the new flag with each of the following programs, here is what happens:
- anvi-setup-kegg-data (only applies when used with the
--download-from-keggoption): anvi'o will estimate bit score thresholds for each nt-KO. If there is more than one sequence belonging to the nt-KO family in KEGG GENES, it will use all of them to generate a new pHMM; otherwise, it will use the pHMM included in the KEGG KOfam database. The new models and thresholds are stored in the KEGG data directory for later use. See more details here. - anvi-run-kegg-kofams: Any nt-KOs that match to gene sequences with a high enough bit score will be included as annotations in your contigs database.
- anvi-estimate-metabolism: Any annotated nt-KOs that participate in KEGG modules will be included in the completeness and copy number calculations.
If you want more context for why this option exists, feel free to check out this paper in which we benchmarked
anvi-run-kegg-kofams. A feature implemented by Iva Veseli (#2237), with debugging help from Benjamin Coltman (#2471, #2336). - anvi-setup-kegg-data (only applies when used with the
-
Other improvements include better memory performance for anvi-estimate-metabolism when it is run in
--metagenome-mode(#2229 by Iva Veseli), the ability to estimate metabolism for gene cluster bins in a pangenome (#2177 by Iva Veseli), and more.
HMM-based annotations work better for large datasets
Thanks to Benjamin Luke Coltman, with assistance from Iva Veseli, all annotation programs that use HMMER under the hood now have improved performance, resulting in fewer memory issues with big datasets in #2423, #2438, and #2469. HMMER's e-value calculations will also now be consistent regardless of how many threads are used to process the data (#2447). And finally, anvi-run-kegg-kofams includes an option to override HMMER's default reporting thresholds, which can eliminate good hits that have high e-values simply due to a huge database size (#2448).
Tools to analyze metabolic networks
This release introduces tools that leverage KEGG Pathway maps to analyze metabolic networks, which are stored in anvi'o databases as 'reaction networks'. anvi-draw-kegg-pathways displays enzymatic capacities using the extensive map database. A new pathway walker algorithm finds reaction paths through the network data underlying the maps, and anvi-predict-metabolic-exchanges compares these paths across genomes to identify potential metabolic exchanges in communities.
Visualizing Metabolic Pathways
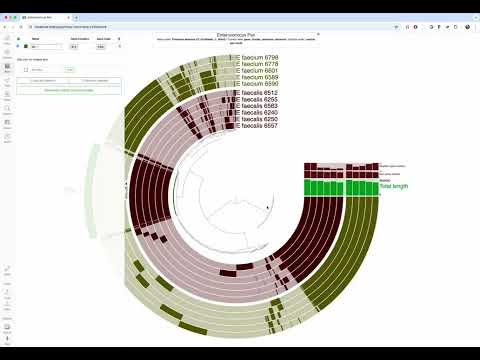
anvi-draw-kegg-pathways (#2485 by Samuel Miller) enables the user to analyze genomic data from contigs or pan databases in the context of metabolic maps. Beyond the simplest application to a single genome, this tool can process groups of genomes, showing, for example, how collections of strains representing two distinct taxa in a single genus differ in their ability to biosynthesize key cofactors. Here is an example of differences between Enterococcus faecalis and Enterococcus faecium in folate biosynthesis:

A variety of options are available to tailor output to the user's needs, including:
-
Biologically informative output directory structure with
--name-filesand--categorize-files(the map above would be stored atMetabolism > Metabolism_of_cofactors_and_vitamins > kos_00790_Folate_biosynthesis.pdf). -
Categorical and continuous color scheme options. The following maps compare the genomes of six Enterococcus faecalis strains stored in contigs databases. The maps were produced with
--colormap plasma 0.1 0.9, which specifies the "plasma" colormap from its 10th to 90th percentile to avoid the lightest and darkest colors, and--reverse-overlay, which renders the bottom global metabolism map in a way that emphasizes the reactions that are not shared by all the genomes, by drawing their lines on top. (Open the image in a new tab to see the extremely high resolution of maps.) -
"Map grid" that displays maps for individual databases alongside the comparison map. The following grid of the "Pentose and glucuronate interconversions" map compares the genomes of twelve Enterococcus faecalis and faecium strains stored in a pan database. The first map shows the number of genomes encoding enzymes for each reaction, and the other maps show the enzymatic capacity of each genome, enabling detailed investigation of metabolic differences. See an example visualization here.
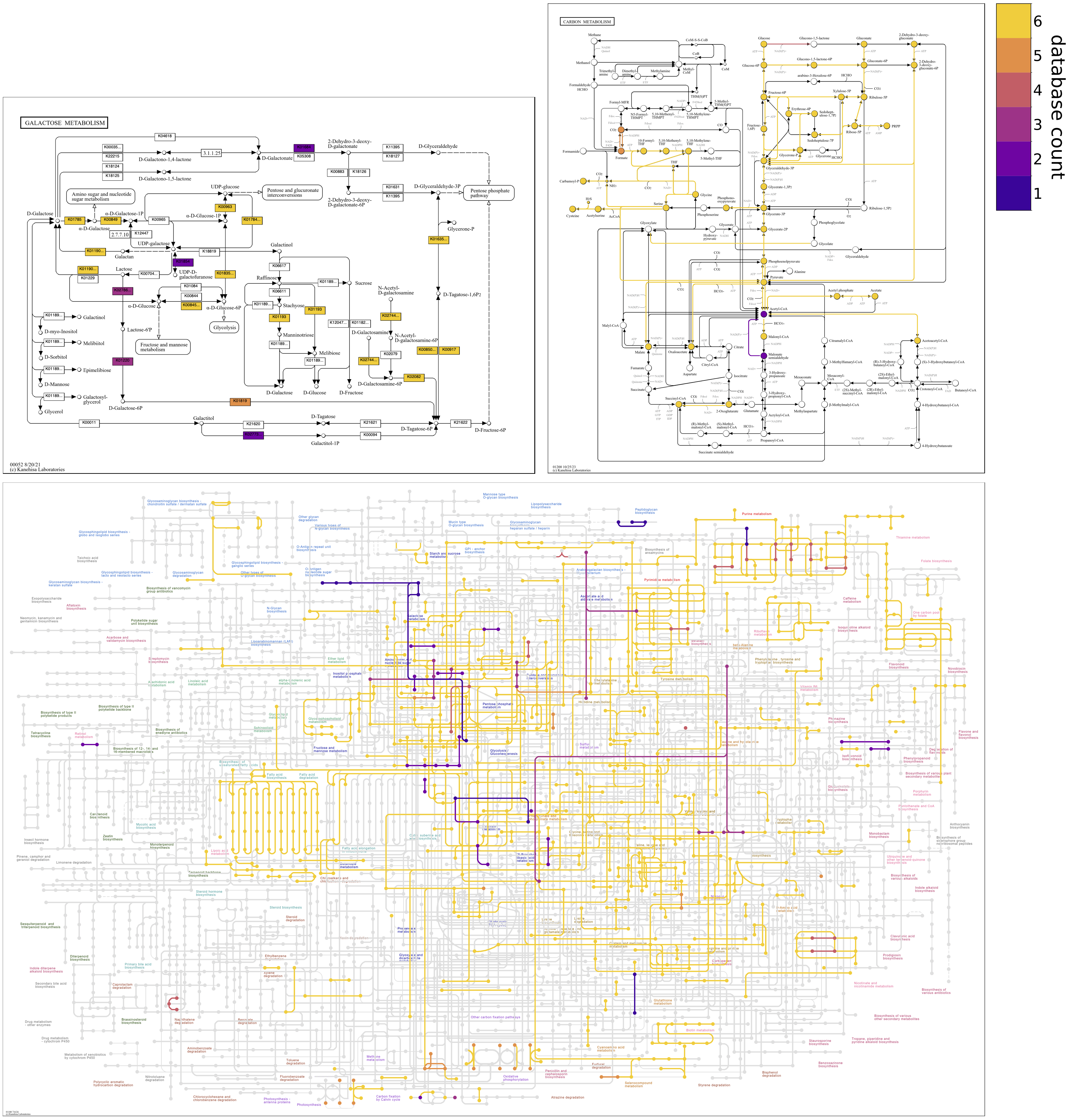
Identifying reaction chains
Associated with each map is a "KGML" (XML) file of metabolic network data. The new anvi'o KGML walker implemented by Samuel Miller traverses chains of reactions linking compounds in the parsed files. Given genomic data from contigs databases, the walker finds metabolic routes available to organisms.
Among other capabilities, the algorithm can extend reaction chains through gaps for which there is no evidence from genomic enzyme annotations. A new gap filling algorithm proposes gene annotations or re-annotations to fill these gaps on the basis of gene synteny and alternative annotations from neighboring genes in the metabolic network. This opens doors to additional capabilities in anvi'o, such as predicting metabolic interactions.
Predicting metabolic interactions
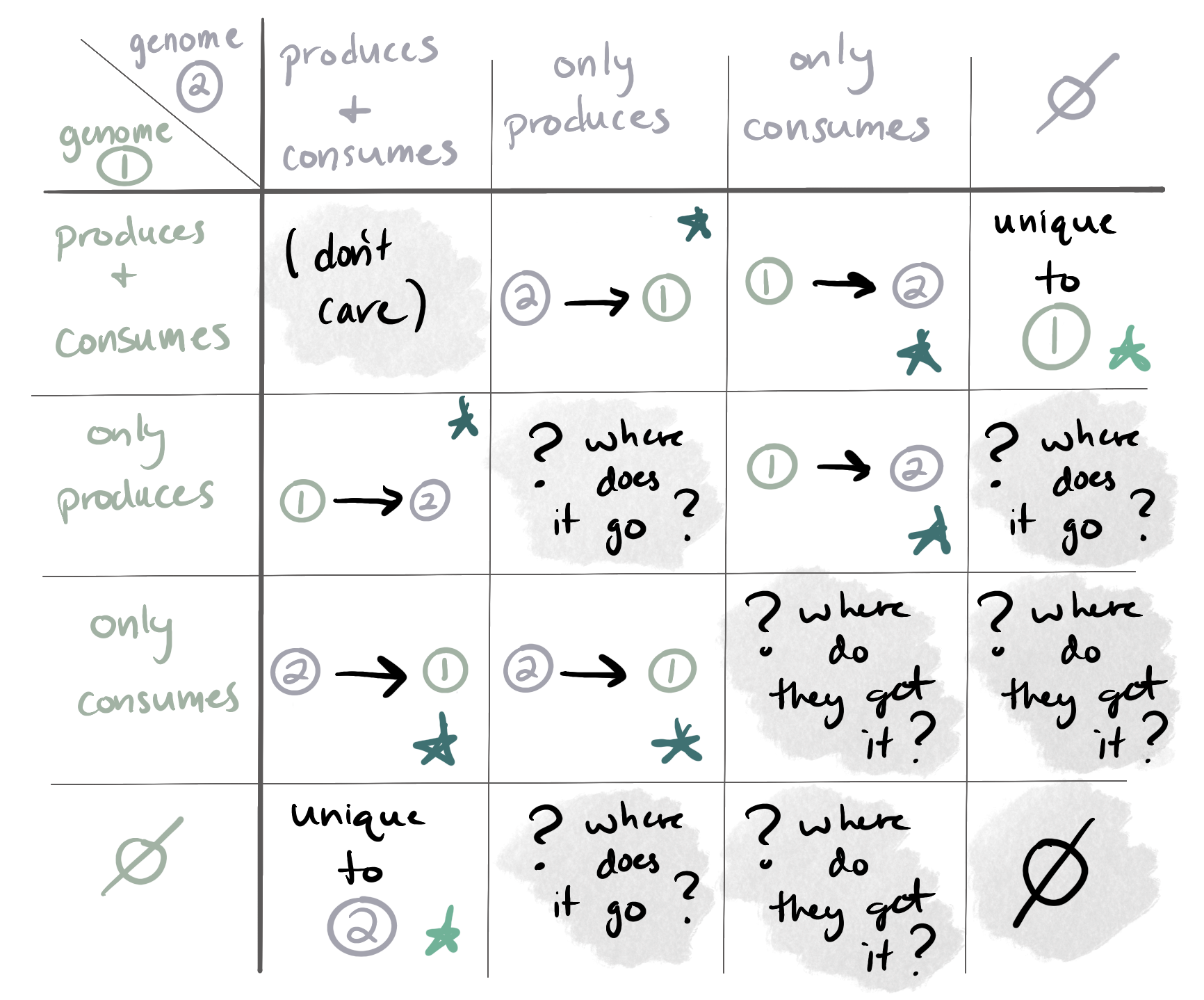
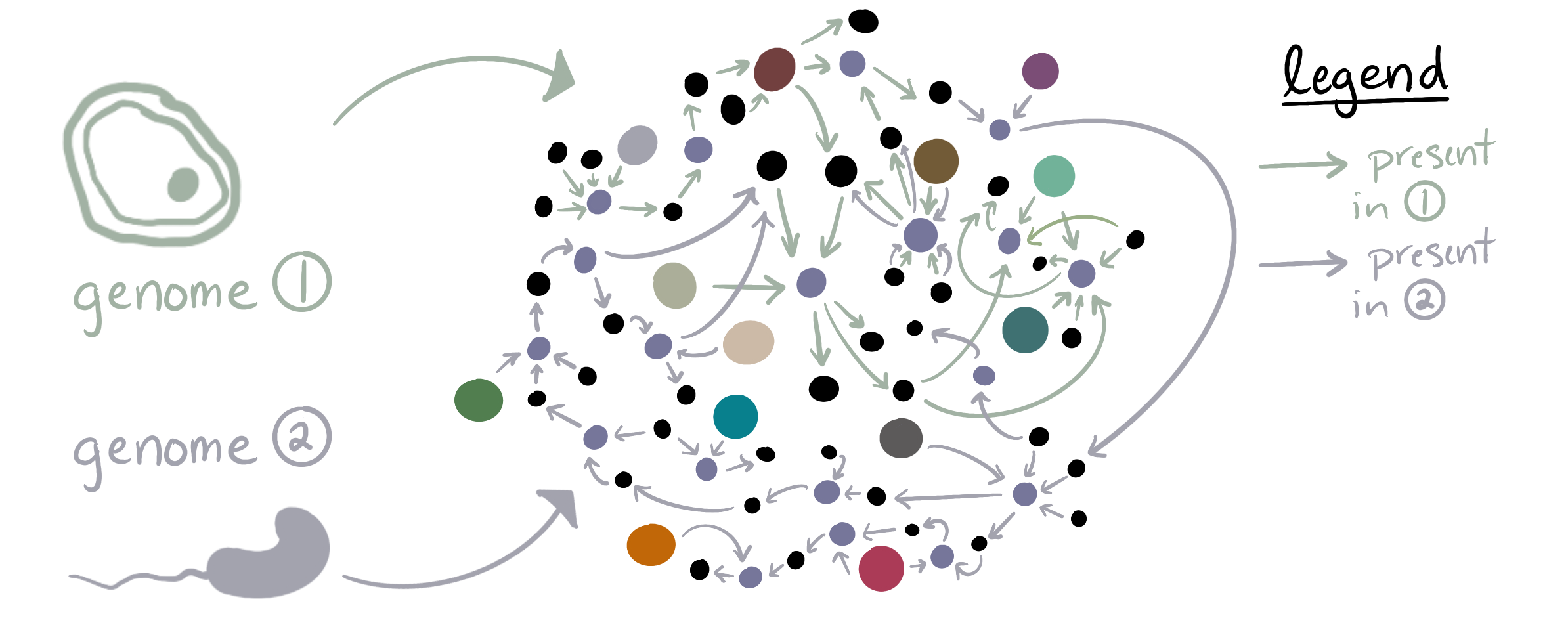
anvi-predict-metabolic-exchanges (#2455 by Iva Veseli) allows the user to predict potentially-exchanged metabolites between pairs of organisms. It does so by examining their reaction networks and looking for (1) compounds that can be produced by only one of the organisms and consumed (as a substrate) by the other, and (2) compounds that can be consumed by only one of the organisms and produced by the other.
{kind=link}
The program leverages two strategies to make these predictions. First, the KGML Walker algorithm to walk over each KEGG Pathway Map and identify potentially-exchanged compounds with reaction chains as evidence for the likelihood of the exchange. Second, it looks across the entire reaction networks of both organisms to identify potential transfer points; these predictions do not have evidence associated with them, but they can include metabolites that are not included in KEGG Pathway Maps. You can find more technical details in the program page here.
anvi-predict-metabolic-exchanges supports multiple pairwise genome comparisons via the external-genomes artifact. It can run either all-vs-all comparisons or specific pairwise comparisons as described in a genome-pairs artifact.
The metagenomics workflow now works with long-read data
The anvi'o workflow for metagenomics has been updated and includes several new features/options:
-
Long read support in the samples-txt. The samples-txt file can include short and long read samples. The columns
r1andr2are used for samples of paired-end reads, and the columnlrfor samples of long reads. You can also combine the read types for a single sample. Here is an example:sample r1 r2 lr sample_01 /path/r1 /path/r2 /path/lr sample_02 /path/r1 /path/r2 sample_03 /path/lr -
Long read assembly and mapping tools. We included (meta)Flye as an option for long read assembly, and minimap2 for long read mapping. For a given sample with both read types, the workflow performs two separate assemblies.
-
Conda environment for assembly and mapping rules in the workflow. You can now provide either the name of an existing conda environment or the path to a conda YAML file for each assembly and mapping rule.
If you want more information about the changes made to the workflow, including minor improvements, check out the pull-request #2479.
A completely redesigned interactive interface
Thanks to Metehan Sever's efforts, the interactive interface of anvi'o has been completely redone for a more organized, modern look. In addition to its new look, it also has additional functionality to edit the color of many bins at once (#2410), benefit from a better search functionality (#2377), much better visualization of and branch support display for phylogenetic trees with support values (#2278, #2208), and more.
Please see https://anvio.org for most up-to-date installation instructions, documentation, and how to reach out to us on Discord for questions.
Since the release of v9, we had a few hot fixes for a minor bugs:
- @jnesme identified an
anvi-setup-pdb-databasedata directory bug in #2521. - @EricDeveaud identified an
anvi-setup-interacdomedata directory bug in #2533
The package below includes all these fixes.