Selecting compounds for docking
Whilst it is possible to get access to vast numbers of molecules, simply plugging one of these vast libraries into docking engine is a huge waste of resources and time since many of the compounds may be unattractive as a starting point (High Molecular Weight, extreme LogP, high TPSA, presence of reactive, or assay interfering groups).
In addition, since the docking tools often only give a 10 to 20-fold enrichment you will end up having to sift through thousands of results. It is far better to employ a series of simple filters to remove undesirable compounds before undertaking the time-consuming and resource intensive docking study.
There are now many publicly available sources of molecules a few are listed below.
In addition, there are tools that allow the user to enumerate large libraries of compounds such as SmiLab, which enumerates combinatorial libraries at rates of approximately 9,000,000 molecules per minute on fast computers.
DataWarrior is a free application for visualisation, filtering and analysis of chemical datasets. Most of DataWarrior's functionality is described in detail in its user manual. DataWarrior installers for Linux, Macintosh and Windows can be downloaded from the download page. This tutorial will give an example of a workflow that might be used to selecting molecules for a docking run. The example chosen is actually from the Open Source Malaria project were OSM-S-106 is a novel inhibitor of PfηCarbonic Anhydrase, but the strategy would be applicable elsewhere.
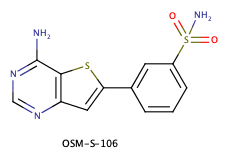
Since carbonic anhydrase has a zinc atom in the active site it is reasonable to assume that the sulphonamide binds to the zinc as is found in this example. A reasonable strategy might be to dock a range of aryl sulphonamides to try and identify interesting novel ligands.
A search of ChEMBL identifies over 110,000 benzenesulphonamides, these were downloaded as a single sdf file. Open DataWarrior and then from the File choose "Open", navigate to the downloaded sdf file and import it.
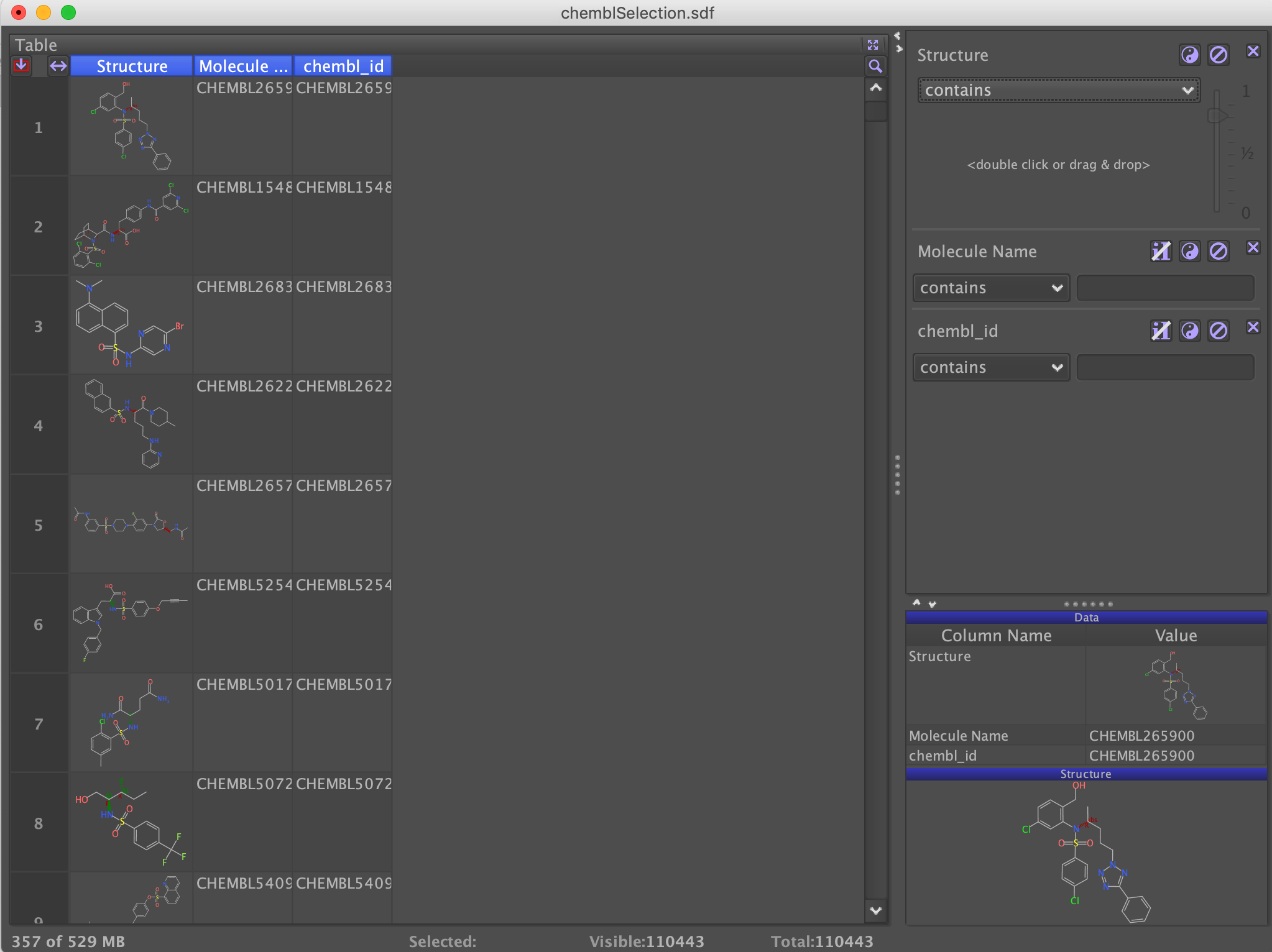
You may have various 2D and 3D viewer panes open, you can close these and open the table view as shown above. The number of molecules displayed in the table is shown at the bottom of the table, in this case 110443. It is possible that the input structures contain some salts or hydrates, from the "From Chemical Structure" menu select "Add Largest Fragment". This removes counter ions from salts, removes water molecules etc. The cleaned largest structure is then written into a new structure column. If the option Neutralize charges is selected, then DataWarrior tries to remove charges to neutralize the overall molecule.
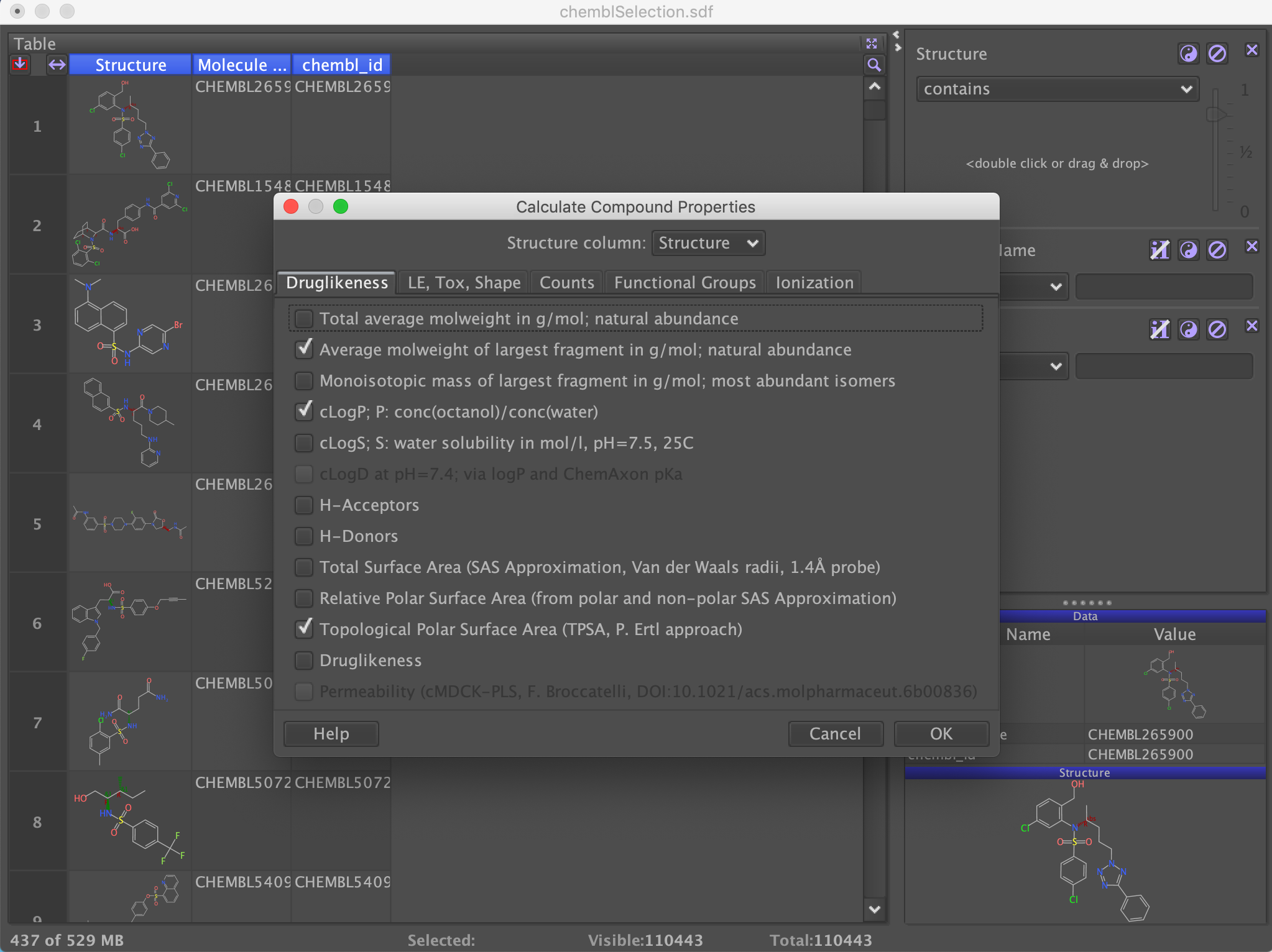
We want to filter these molecules based on a variety of calculated properties such as Mol Weight, LogP etc. From the top menu bar select "Chemistry" then select "Calculate Properties" from the "From Chemical Structure" menu item. These gives a dialog with the option to calculate a variety physicochemical properties and descriptors. Choose a selection of descriptors and click OK.
As you can see in the image there are a number of things we can use to exclude molecules, a number contain undesirable functional groups. Some are very molecular weight, you probably want to aim for <400, there are some that have extreme values for calculated logP you probably want between 0 and 4, some also have very high polar surface area, compounds with TPSA > 110 tend to have poor oral absorption. There is a report on the properties of "drug-like" molecules here.
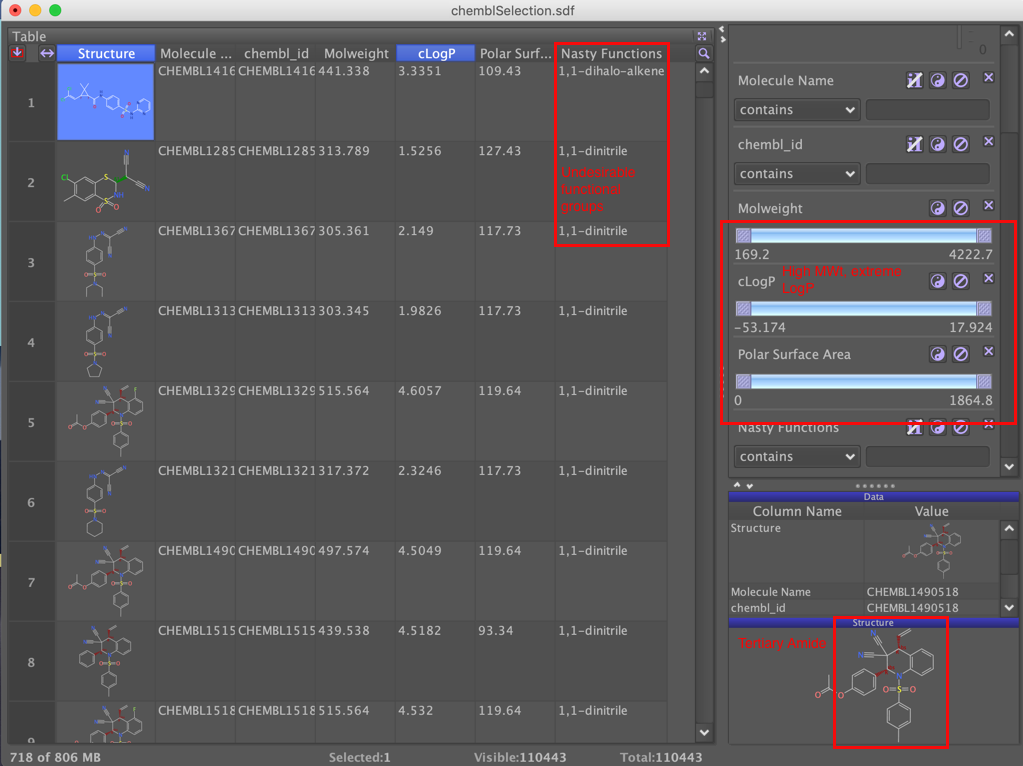
We can also exclude tertiary sulphonamides since these are unlikely to be able to bind to the zinc
This filters the selection down from over 110,000 to around 12,000. You can further refine the list by looking at other descriptors or Delete invisible rows (from data menu) and then choose "Cluster compounds" from the Chemistry menu.
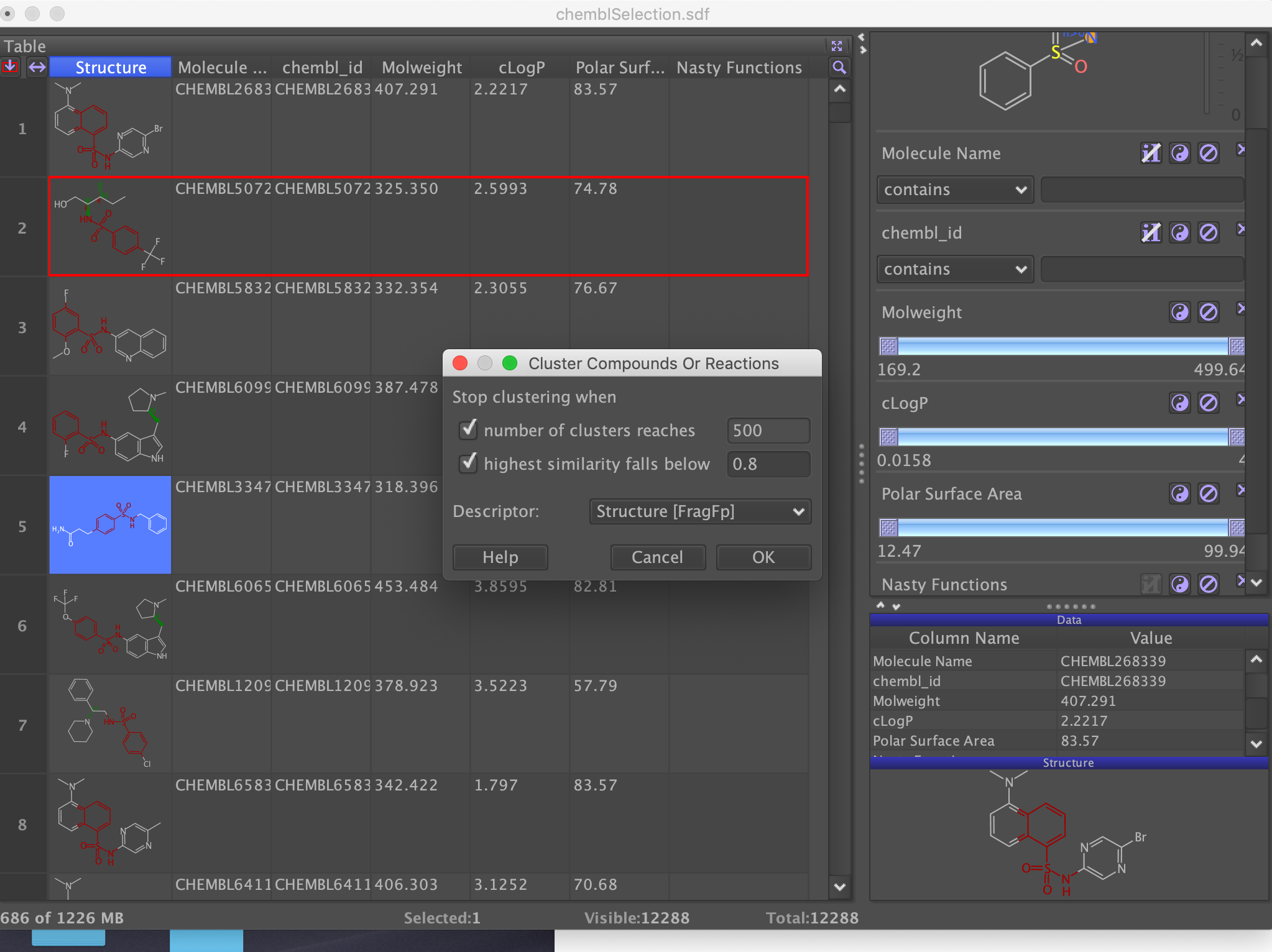
This may take a little while depending on the number of molecules, but you can then select interesting examples from each cluster or you can simply choose the "Is representative" option. Then choose "Save Special" from the File menu to save as an sdf file. This is the file that will be used as the input for docking.