Refer to our GitHub: Py4LongReadRNASeq
Authors: Zhongshan Cheng1, Jia-Hua “George” Qu2, Nick Peterson3, Meghan Carr4
Affiliations:
- Center for Applied Bioinformatics, St. Jude Children’s Research Hospital
- Host-Microbe Interactions, St. Jude Children’s Research Hospital
- Host-Microbe Interactions, St. Jude Graduate School (Ph.D. track)
- Student, CodeCrew Coding School
This repository documents the steps for running a long-read RNA-Seq analysis pipeline integrating PacBio Iso-Seq, IsoQuant, and our modified TAGET workflow. The pipeline enables isoform-level and gene-level quantification, followed by differential expression (DE) analysis with DESeq2.
-
Create a dedicated conda environment:
conda create -n pacbio python=3.8.20 conda activate pacbio
-
Install required packages:
python=3.8.20 samtools=1.10 openssl=1.1.1w minimap2=2.26 hisat2=2.2.1 pandas=1.3.5 numpy=1.21.6
-
Export environment if needed:
conda env export -n pacbio > environment-pacbio.yaml
-
Download example BAM:
wget https://downloads.pacbcloud.com/public/dataset/Melanoma2019_IsoSeq/FullLengthReads/flnc.bam
-
Split BAM into per-sample files. Example:
samtools view transcripts.bam -h | perl ... > sample.sam bash PreparePacbioClusteredBAM2TAGET.sh sample.sam sampleID
- Download hg38 reference and annotation (GENCODE or Ensembl).
-
Convert BAM to FASTA per sample.
-
Generate TPM/CPM tables:
conda activate pacbio python Py4LongReadRNASeq/DetermineExp4ClusteredReads.py -f input.fasta -o output_for_TAGET.tpm.txt
-
Update config file with CPM paths.
-
Run TAGET refinement:
conda activate pacbio python RefinedLongReadMappingAndQuantification.py -c config
-
Merge isoform results:
conda activate pacbio python TransAnnotMerge.py -c merge.config -o outputdir -m FLC
Outputs include: gene.exp, transcript.exp, and supporting files.
-
Install isoquant:
conda create -c bioconda -n isoquant python=3.8 isoquant=3.7.1 conda activate isoquant conda env export -n isoquant > environment-isoquant.yaml
-
Download the fastq file
wget https://downloads.pacbcloud.com/public/dataset/Melanoma2019_IsoSeq/FullLengthReads/flnc.fastq
-
Run isoquant on FASTQ:
isoquant.py --reference hg38.fa --genedb hg38.gtf --fastq flnc.fastq \ --data_type pacbio_ccs -o output_isoquant
This step also generated the new gft file, the IsoQuant-derived GTFs incorporating novel transcripts.
-
Supports running using Singularity containers:
pbskera,isoseq,pbmm2,pbpigeon. -
Includes alignment, isoquant refinement, pigeon classification, and annotation processing.
-
This optional step is to generate results to compare the functions of the PacBio Standard Pipeline and our modified TAGET pipeline.
-
Either the PacBio Standard Pipeline or our modified TAGET pipeline produces at least two output results, including gene.exp and transcript.exp.
-
To save time, we performed the analysis in the St. Jude HPC this time.
module load parallel/20240222 export USER=zcheng export HG38=/research/rgs01/applications/hpcf/authorized_apps/hartwell/Automation/REF/Kinnex-IsoSeq/RefGenomes/Human_hg38_Gencode_v39 export REFS=/research/rgs01/applications/hpcf/authorized_apps/hartwell/Automation/REF/Kinnex-IsoSeq mkdir -p /scratch_space/$USER/KinnexBulkIsoSeqAnalysis cd /scratch_space/$USER/KinnexBulkIsoSeqAnalysis singularity pull docker://quay.io/biocontainers/pbskera:1.4.0--hdfd78af_0 singularity pull docker://quay.io/biocontainers/lima:2.13.0--h9ee0642_0 singularity pull docker://quay.io/biocontainers/isoseq:4.3.0--h9ee0642_0 singularity pull docker://quay.io/pacbio/pbmm2:1.17.0_build1 singularity pull docker://quay.io/biocontainers/pbfusion:0.5.1--hdfd78af_0 # Run ISOSEQ REFINE # Don't need to run this step, because we have downloaded the flnc.bam in the beginning. # wget https://downloads.pacbcloud.com/public/dataset/Melanoma2019_IsoSeq/FullLengthReads/flnc.bam # If we don't have the refined file, flnc.bam, the user needs to run this code. singularity run -B $PWD -B $REFS -B $HG38 docker://quay.io/biocontainers/isoseq:4.3.0--h9ee0642_0 \ isoseq refine --require-polya -j 16 \ 02_COLO829Pacbio.{1}.fl.{2}.bam \ $REFS/bulkRNA/02_lima-primers/IsoSeq_v2_primers_12.fasta \ 03_COLO829Pacbio.{1}.flnc.bam ### x.sh # Use previously generated or downloaded flnc BAM; https://downloads.pacbcloud.com/public/dataset/Melanoma2019_IsoSeq/ https://github.com/RhettRautsaw/StJude_PacBio-WDL-tutorial/blob/main/Kinnex_IsoSeq_Pipelines/KinnexBulkIsoSeq.lsf # extract reads for each sample; # Search and extract the reads identified in a certain sample, and generate the subset sam file. export sampleID=$1; samtools view flnc.bam -h |perl -anE 'if (/^\@/){print}else{print if /$ENV{sampleID}/} ' |samtools view -Sb - -o $sampleID.bam; samtools index $sampleID.bam # put the above into x.sh to run it for each sample in the cluster; bsub_Grace_Next -n 1 -m 20 "bash x.sh m54019_190120_021709" bsub_Grace_Next -n 1 -m 20 "bash x.sh m54026_190120_000756" bsub_Grace_Next -n 1 -m 20 "sleep 40s;bash x.sh m54119_190202_095143" bsub_Grace_Next -n 1 -m 20 "sleep 100s;bash x.sh m54119_190203_061153" bsub_Grace_Next -n 1 -m 20 "sleep 200s;bash x.sh m54119_190131_171128" bsub_Grace_Next -n 1 -m 20 "sleep 300s;bash x.sh m54119_190201_133141" ### y.sh export USER=zcheng export HG38=/research/rgs01/applications/hpcf/authorized_apps/hartwell/Automation/REF/Kinnex-IsoSeq/RefGenomes/Human_hg38_Gencode_v39 export REFS=/research/rgs01/applications/hpcf/authorized_apps/hartwell/Automation/REF/Kinnex-IsoSeq realpath $1.bam > 03_COLO829Pacbio.$1.flnc.fofn # Run PBMM2 singularity run -B $PWD -B $REFS -B $HG38 docker://quay.io/pacbio/pbmm2:1.17.0_build1 \ pbmm2 align -j 16 --preset ISOSEQ --sort \ $HG38/human_GRCh38_no_alt_analysis_set.fasta \ 03_COLO829Pacbio.$1.flnc.fofn \ 04_COLO829Pacbio.$1.align.bam #put the above codes into y.sh to run it for each sample; #Run all samples using the y.sh ls m*bam | perl -pe 's/\.bam//' | perl -ane 'chomp;`bsub_Grace_Next -n 1 -m 100 "bash y.sh $_"`' realpath 04_COLO829Pacbio.*.align.bam > 04_COLO829Pacbio.align.fofn perl -pe 's/.*kinnex.//g' 04_COLO829Pacbio.align.fofn | perl -pe 's/.align.bam//g' > 04_COLO829Pacbio.align.labels singularity run -B $PWD -B $REFS -B $HG38 docker://quay.io/pacbio/pb_wdl_base:build3 \ python $REFS/isoquant_generateYAML.py -b 04_COLO829Pacbio.align.fofn -l 04_COLO829Pacbio.align.labels -e 05_COLO829Pacbio.isoquant -o 05_isoquant4COLO829.yaml rm 05_COLO829Pacbio.isoquant -rf singularity run -B $PWD -B $REFS -B $HG38 docker://quay.io/biocontainers/isoquant:3.6.3--hdfd78af_0 \ isoquant.py -t 64 -d pacbio --yaml 05_isoquant4COLO829.yaml \ -r $HG38/human_GRCh38_no_alt_analysis_set.fasta \ -g $HG38/gencode.v39.annotation.sorted.gtf.db --complete_genedb \ -o 05_COLO829Pacbio.isoquant \ --sqanti_output ln -s 05_COLO829Pacbio.isoquant/05_COLO829Pacbio.isoquant/05_COLO829Pacbio.isoquant.* . singularity run -B $PWD -B $REFS -B $HG38 docker://quay.io/pacbio/pb_wdl_base:build3 \ python $REFS/isoquant2pigeon.py \ --gtf 05_COLO829Pacbio.isoquant.transcript_models.gtf \ --tsv 05_COLO829Pacbio.isoquant.transcript_model_grouped_counts.tsv \ --output 06_COLO829Pacbio.pigeon.transcript_model_grouped_counts.csv singularity run -B $PWD -B $REFS -B $HG38 docker://quay.io/biocontainers/pbpigeon:1.4.0--h9948957_0 \ pigeon classify -j 64 -o 06_COLO829Pacbio.pigeon \ 05_COLO829Pacbio.isoquant.transcript_models.gtf \ $HG38/gencode.v39.annotation.sorted.gtf \ $HG38/human_GRCh38_no_alt_analysis_set.fasta \ --flnc 06_COLO829Pacbio.pigeon.transcript_model_grouped_counts.csv \ --cage-peak $HG38/refTSS_v3.3_human_coordinate.hg38.sorted.bed \ --poly-a $HG38/polyA.list.txt \ --coverage $HG38/intropolis.v1.hg19_with_liftover_to_hg38.tsv.min_count_10.modified2.sorted.tsv singularity run -B $PWD -B $REFS -B $HG38 docker://quay.io/biocontainers/pbpigeon:1.4.0--h9948957_0 \ pigeon report -j 64 06_COLO829Pacbio.pigeon_classification.txt 06_COLO829Pacbio.pigeon_classification.report.txt cat 05_COLO829Pacbio.isoquant.extended_annotation.gtf | grep transcript | perl -pe 's/.*gene_id.{2}(ENSG\d+\.\d+).; transcript_id .(ENST\d+\.\d+).;.*; .*gene_name "(\S+)".*transcript_name "(\S+)".*/$1\t$2\t$3\t$4/' |sort -u |grep gene_id -v >ensembl_gene2tx.txt #Clean the file headers; perl -i.bak -pe 's/\/[^\t]+\/04_COLO829Pacbio.//g' 05_COLO829Pacbio.isoquant.transcript_model_grouped_counts.tsv perl -i.bak -pe 's/\/[^\t]+\/04_COLO829Pacbio.//g' 05_COLO829Pacbio.isoquant.gene_grouped_counts.tsv ln -s 05_COLO829Pacbio.isoquant.transcript_model_grouped_counts.tsv 07_isoquant.isoforms4COLO829.matrix ln -s 05_COLO829Pacbio.isoquant.gene_grouped_counts.tsv 07_isoquant.genes4COLO829.matrix #Sample matrix, including sample grp and sample ids; #COLO8299T m54026_190120_000756 #COLO8299T m54119_190202_095143 #COLO8299T m54119_190203_061153 #COLO829BL m54019_190120_021709 #COLO829BL m54119_190131_171128 #COLO829BL m54119_190201_133141
Performed with DESeq2 (via Trinity Singularity image). Below are the detailed steps for both PacBio and TAGET pipelines.
-
Annotate transcript IDs with gene names:
vlookup 07_isoquant.isoforms4COLO829.matrix 1 ensembl_gene2tx.txt 2 3,4 y | \ perl -ane '$F[0]=$F[-1] unless /NaN/;print join("\t",@F[0..($#F-2)]),"\n";' \ > 07_isoquant.isoforms4COLO829.matrix.new
-
Run DESeq2:
singularity run -B $PWD -B $REFS -B $HG38 \ docker://quay.io/biocontainers/trinity:2.15.2--pl5321h077b44d_3 \ run_DE_analysis.pl \ --matrix 07_isoquant.isoforms4COLO829.matrix.new \ --method DESeq2 \ --samples_file 07_samples.matrix4COLO829.txt \ --output 07_deseq2.isoforms
-
Extract top 20 isoforms:
head -n21 07_deseq2.isoforms/07_isoquant.isoforms4COLO829.matrix.new.COLO8299T_vs_COLO829BL.DESeq2.DE_results | \ cut -f1 | tail -n +2 > 07_deseq2.top20.isoforms.new.txt
-
Annotate gene IDs with gene names:
vlookup 07_isoquant.genes4COLO829.matrix 1 ensembl_gene2tx.txt 1 3 y | \ perl -ane '$F[0]=$F[-1] unless /NaN/;print join("\t",@F[0..($#F-1)]),"\n";' | \ SortFileByCols.sh - '-k1,1 -u ' 1 > 07_isoquant.genes4COLO829.matrix.new
-
Run DESeq2:
singularity run -B $PWD -B $REFS -B $HG38 \ docker://quay.io/biocontainers/trinity:2.15.2--pl5321h077b44d_3 \ run_DE_analysis.pl \ --matrix 07_isoquant.genes4COLO829.matrix.new \ --method DESeq2 \ --samples_file 07_samples.matrix4COLO829.txt \ --output 07_deseq2.genes
-
Extract top 20 genes:
head -n21 07_deseq2.genes/07_isoquant.genes4COLO829.matrix.new.COLO8299T_vs_COLO829BL.DESeq2.DE_results | \ cut -f1 | tail -n +2 > 07_deseq2.top20.genes.new.txt
-
Format TAGET transcript expression file:
cat ~/working_scripts/Py4LongReadRNASeq/outputdir/transcript.exp | \ delete_column 1 | \ perl -pe 's/Transcript/#feature_id/' | \ SortFileByCols.sh - '-k1,1 -u ' 1 > 07_TAGET.isoforms4COLO829.matrix.new
-
Run DESeq2:
singularity run -B $PWD -B $REFS -B $HG38 \ docker://quay.io/biocontainers/trinity:2.15.2--pl5321h077b44d_3 \ run_DE_analysis.pl \ --matrix 07_TAGET.isoforms4COLO829.matrix.new \ --method DESeq2 \ --samples_file 07_samples.matrix4COLO829.txt \ --output 07_deseq2.isoforms_TAGET
-
Extract top 20 isoforms:
head -n21 07_deseq2.isoforms_TAGET/07_TAGET.isoforms4COLO829.matrix.new.COLO8299T_vs_COLO829BL.DESeq2.DE_results | \ cut -f1 | tail -n +2 > 07_deseq2.top20.isoforms.TAGET.new.txt
-
Format TAGET gene expression file:
cat ~/working_scripts/Py4LongReadRNASeq/outputdir/gene.exp | \ perl -pe 's/Gene/#feature_id/' | \ SortFileByCols.sh - '-k1,1 -u ' 1 > 07_TAGET.genes4COLO829.matrix.new
-
Run DESeq2:
singularity run -B $PWD -B $REFS -B $HG38 \ docker://quay.io/biocontainers/trinity:2.15.2--pl5321h077b44d_3 \ run_DE_analysis.pl \ --matrix 07_TAGET.genes4COLO829.matrix.new \ --method DESeq2 \ --samples_file 07_samples.matrix4COLO829.txt \ --output 07_deseq2.genes_TAGET
-
Extract top 20 genes:
head -n21 07_deseq2.genes_TAGET/07_TAGET.genes4COLO829.matrix.new.COLO8299T_vs_COLO829BL.DESeq2.DE_results | \ cut -f1 | tail -n +2 > 07_deseq2.top20.genes.TAGET.new.txt
- Both PacBio Standard Pipeline and TAGET-modified Pipeline produce isoform- and gene-level DE results.
- Top isoforms and genes can be extracted for downstream biological interpretation.
- Running both pipelines in parallel enables comparison and validation of results, highlighting concordance as well as novel isoform discoveries.
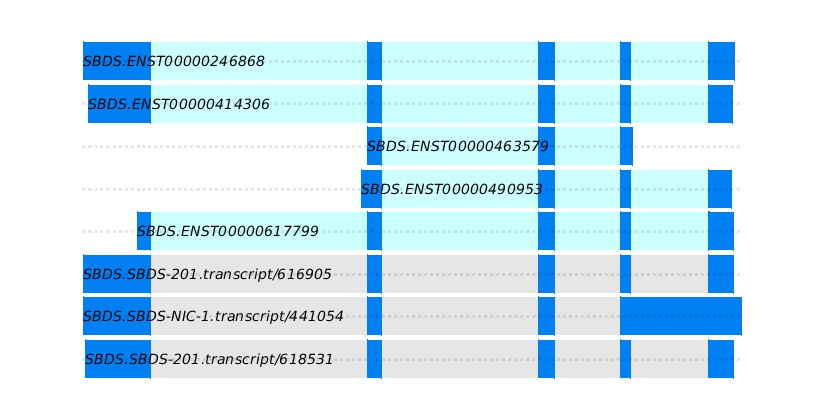
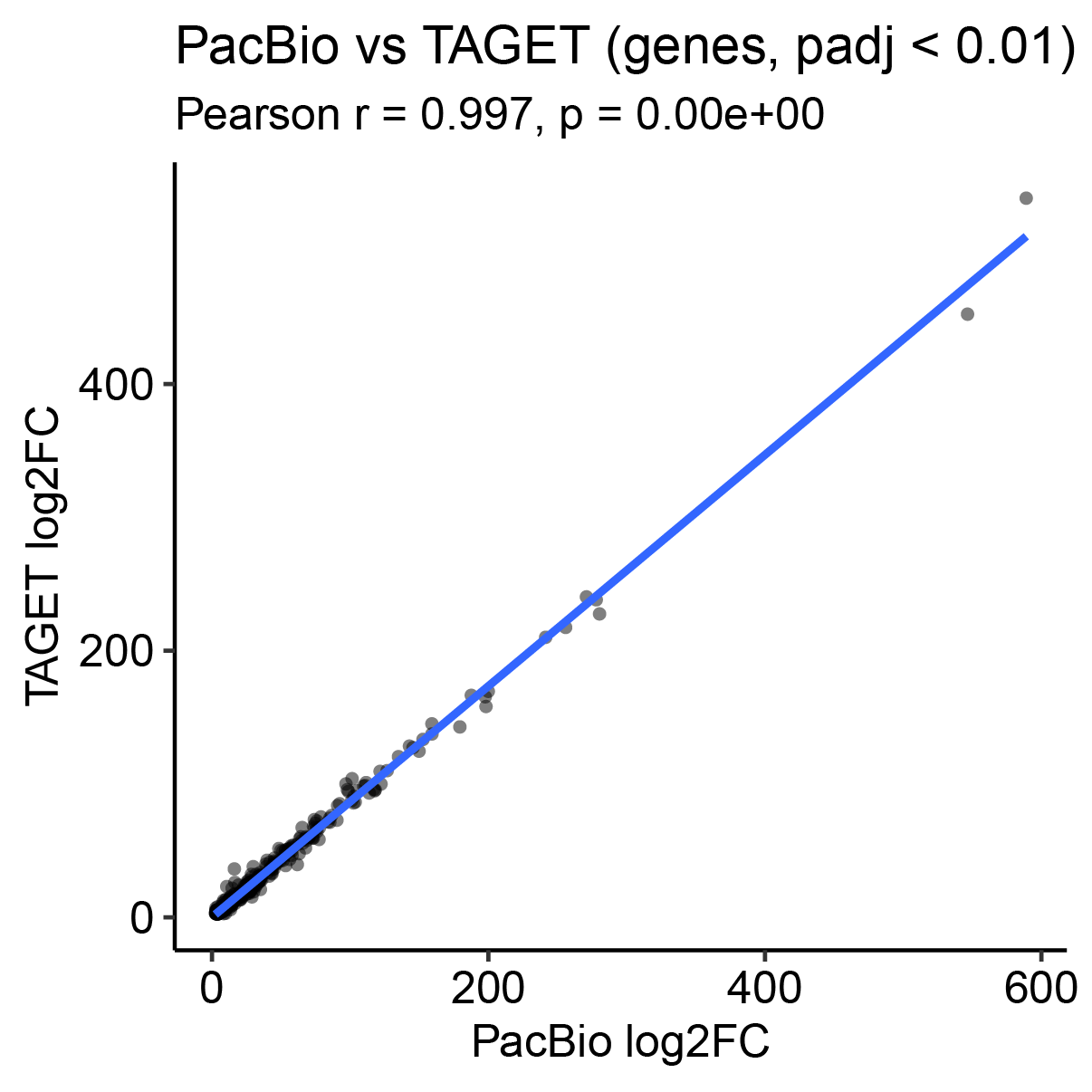
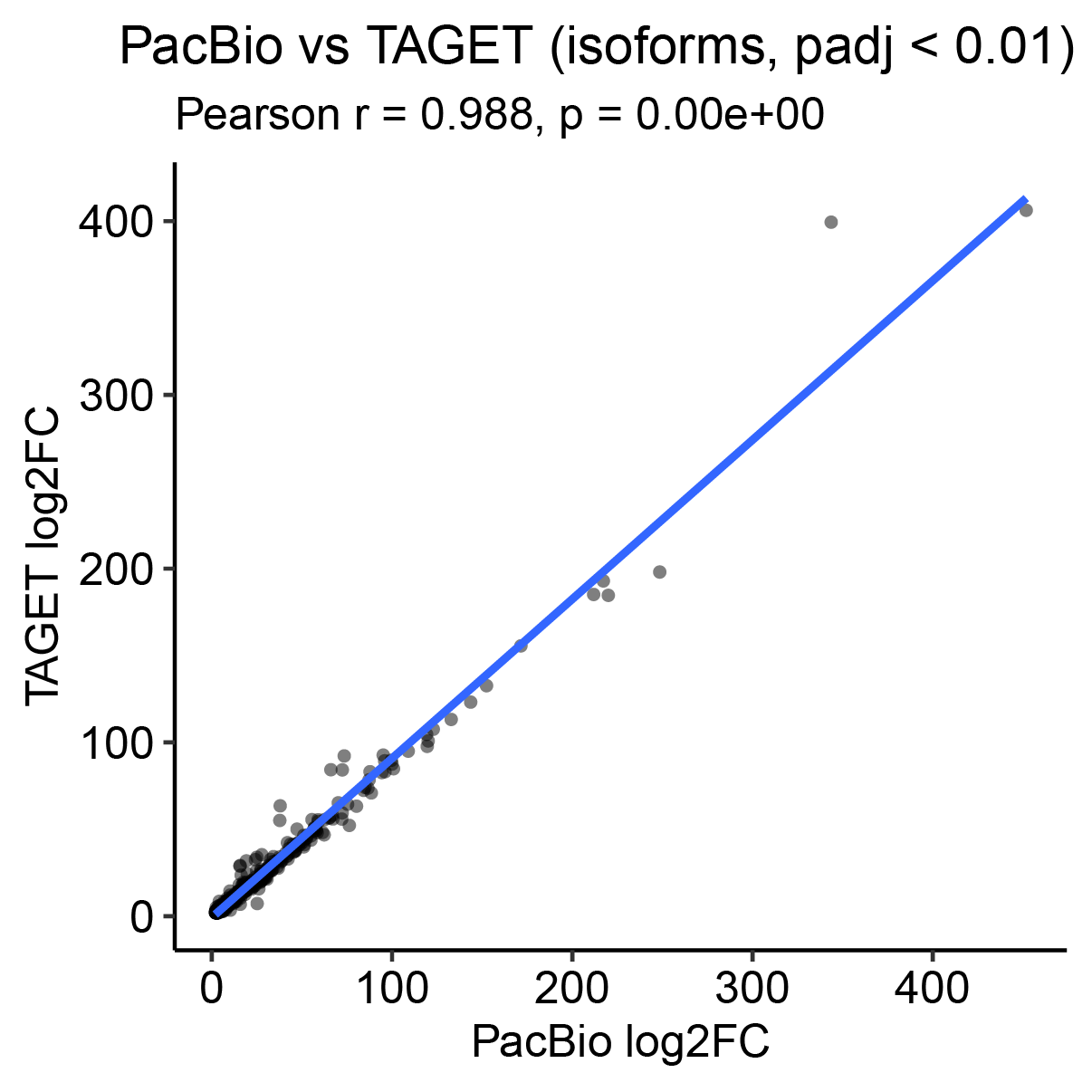
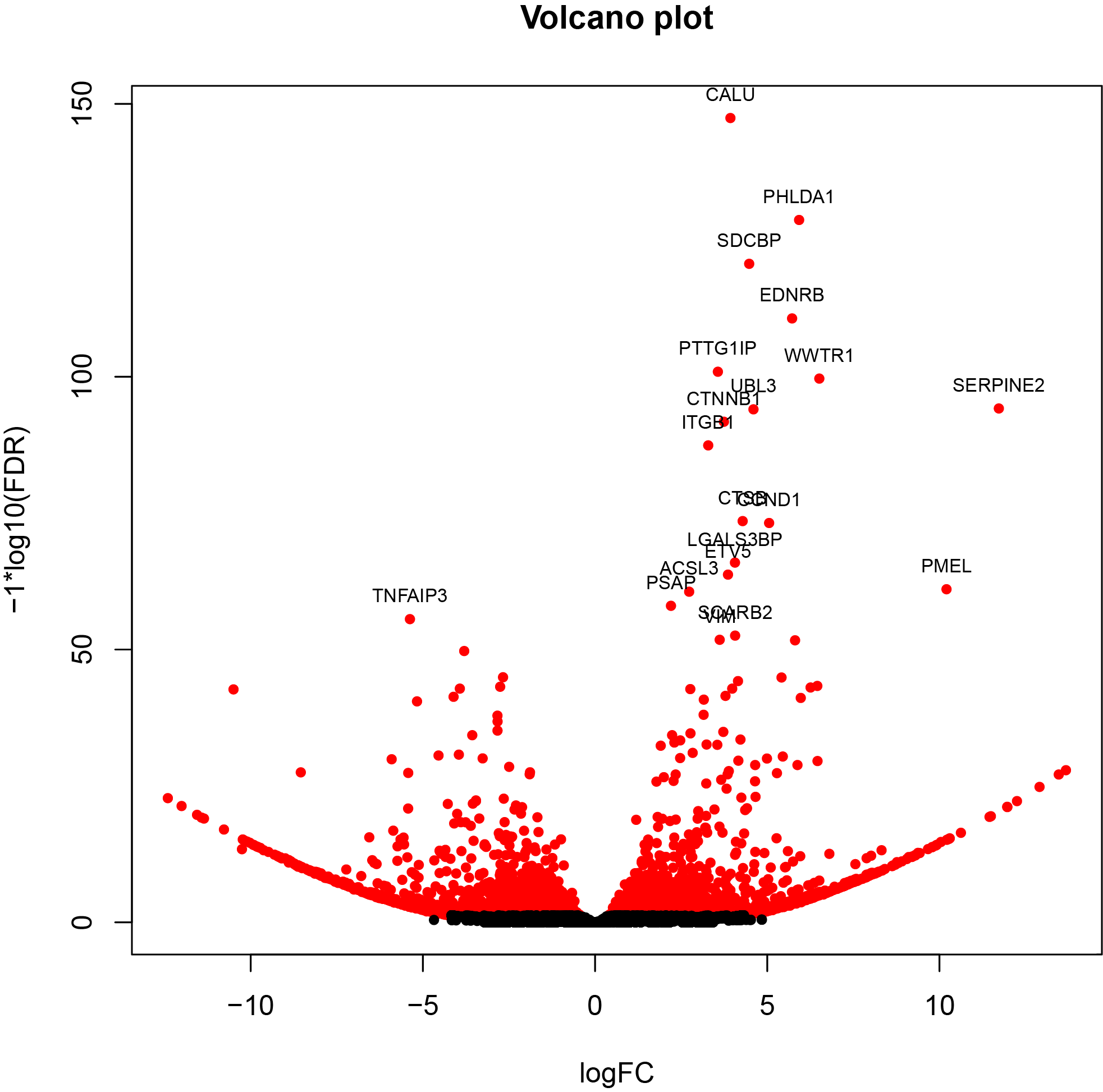
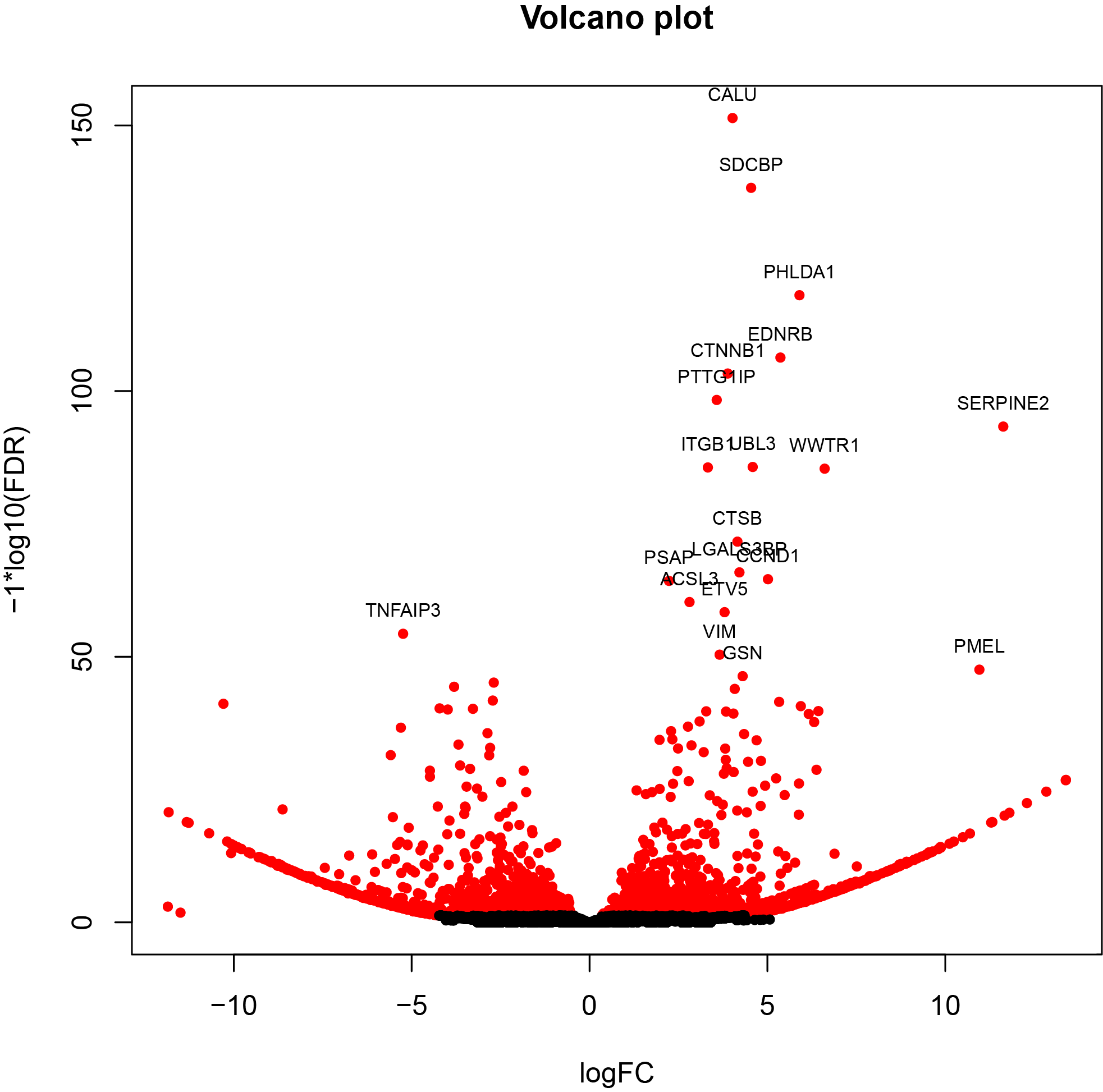
- Ensure reference genome and annotation files are consistent across steps.
- Results can be further rerun using IsoQuant-derived GTFs to incorporate novel transcripts.