02. Project Plan
Leptospirillum ferriphilum is an acidophilic and chemo-lithoautotrophic (meaning that they "make their own food" from chemicals found in minerals) bacteria. L. ferriphilum is one of the most prevalent iron oxidizers and it can survive solely on oxidizing Fe2+ to Fe3+. It is found in acidic and metal-rich environments such as mine drainage and acid rock. L. ferriphilums special niche means that it can be used in biomining where living organisms extract metals from solid material. Biomining is considered more environmentally friendly than regular leaching and L. ferriphilum is thus of great interest. Even though it is an important species much is still unknown about how L. ferriphilum is adapted to its environment. Interesting aspects include metal resistance, oxidative stress and maintaining a neutral pH within the cell in acidic environments. Mapping the genome, transcriptome and proteome of L. ferriphilum is the first step of gaining greater understanding of the physiological processes involved in such adaption. This could among other things help improve the efficiency of biomining (Johnson 2014, Christel et al. 2018).
The their article, Multi-omics Reveals the Lifestyle of the Acidophilic, Mineral-Oxidizing Model Species Leptospirillum ferriphilum, Christel et al. (2018) presents the first high quality, functionally annotated full reference genome for L. ferriphilum. In addition to this the genome was also analysed in combination with transcriptome and proteome data to map different biological processes and metabolic pathways. A differential gene expression and protein concentration analysis was also performed between L. ferriphilum grown in continuous culture and bioleaching culture.
The aim of this project is to recreate some of the analysis performed by Christel et al. (2018).
The analysis is divided in two parts. Firstly the genome will be assembled from long reads obtained by whole genome sequencing using PacBio. The genome will then be annotated and used to investigate synteny. Secondly transcriptomics and differential gene expression analysis will be performed on RNA-seq data obtained by the Illumina TruSeq Stranded total RNA kit (paired reads). For the second analysis two treatments are considered (continuous and bioleaching culture) as mentioned in the introduction. For details on software see the software section in the wiki.
Data: WGS
| Task | Software | Expected Running Time | |
|---|---|---|---|
| 1.1 | Quality control of raw reads + Genome assembly | Canu | ~ 11,5 h (2 cores) |
| 1.2 | Assembly quality assessment | QUAST | < 15 min (1 core) |
| 1.3 | Assembly quality assessment | MUMmerplot | < 5 min (1 core) |
| 1.4 | Structural and functional annotation | Prokka | < 5 min (2 cores) |
| 1.5 | Structural and functional annotation | eggNOGmapper | ~ 1 h (HMM algorithm) |
| 1.6 | Synteny comparison with a closely related genome | blastn |
Data: RNA-Seq
| Task | Software | Expected Running time | |
|---|---|---|---|
| 2.1 | Quality control of raw reads | FastQC | |
| 2.2 | Trimming | Trimmomatic | ~ 15 min per file (2 cores) |
| 2.3 | Quality control after trimming | FastQC | |
| 2.4 | Mapping transcriptome | BWA | ~ 5 h (2 cores) |
| 2.5 | Counting RNA-seq reads | Htseq | ~ 8 h |
| 2.6 | Differential Gene Expression | Deseq2 (R library) |
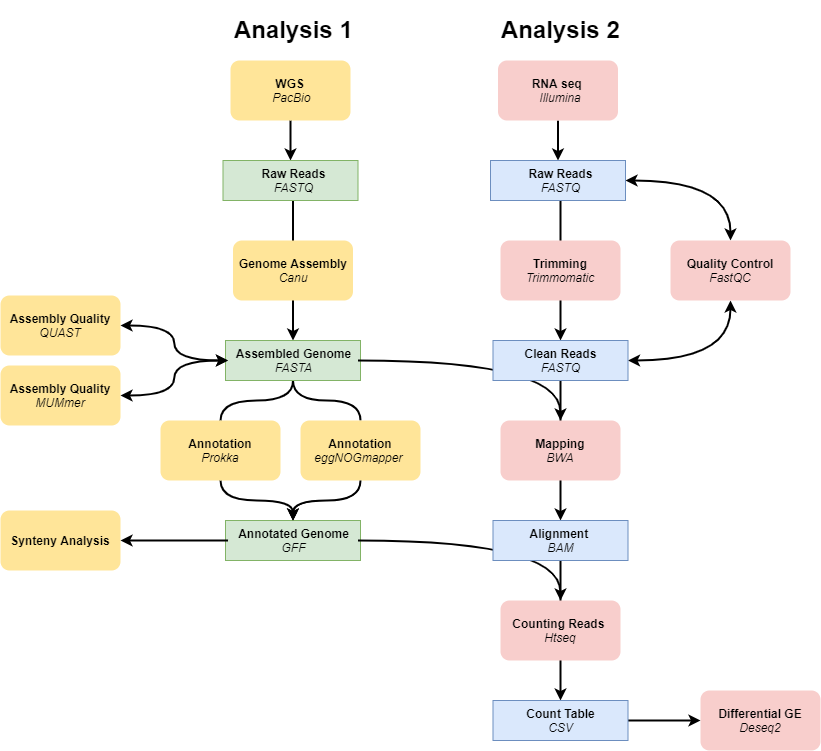
Figure 1. Workflow for analysis 1 and 2. The genome of L. ferriphilum will be assembled and annotated and synteny will be investigated. RNA-seq data will be mapped and a differential gene expression analysis will be performed using the reference genome from analysis 1.
An overview of the timeplan week to week is presented below. The numbers in parenthesis corresponds to the numbers in the methods part of the project plan.
| Week | Tasks Started | Tasks Completed (at the latest) |
|---|---|---|
| 13 | Research topic, understand article and work on project plan | |
| 14 | Genome assembly (1.1) | Research plan |
| 15 | Assembly quality (1.2-1.3), Genome annotation (1.4-1.5) | |
| 16 | RNA-seq preprocessing (2.1-2.3) | Genome assembly and annotation |
| 17 | RNA mapping (2.4) | |
| 18 | Counting RNA-seq reads (2.5), Differential Gene Expression (2.6) | RNA mapping |
| 19 | Synteny (1.6), extra analyses if time | |
| 20 | ||
| 21 | Everything |
The original data used in this project consists of whole genome sequencing by long reads obtained using PacBio and RNA-seq data obtained using Illumina. The original data is stored as FASTQ files. Large files such as the FASTQ files and BAM files will be kept in project folder on UPPMAX where the computationally heavy analyses will be run. Smaller files, code, output and results will be kept in the folder "genome_analysis" on my local machine and GitHub. GitHub will be used as version control and to collect everything related to the project in one place. I will use up to date and correct file formats to store my data. Through each step of the analysis the aim is to, if possible, output a file which can be used as input in the next step without to much modification. To achieve this it will be important to read documentation and specifications carefully to not waste time. Files will be named in a reasonable way so that it's easy to understand what they contain. Filenames of files related to analysis 1 will start with "WGS" and files related to analysis 2 will start with "RNA". The code and data will be separated in different folders and the files to analysis 1 and 2 will be separated in different folders. The overall structure of the directory can be seen in figure 2. During the project this structure might have to be updated and extended to include additional sub folders.
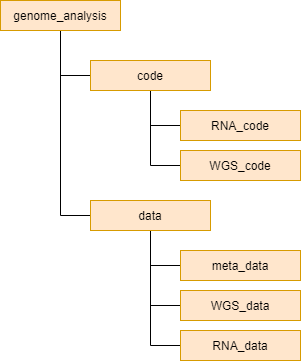
Figure 2. The over all folder structure of the GitHub repository.
Christel S, Herold M, Bellenberg S, El Hajjami M, Buetti-Dinh A, Pivkin IV, Sand W, Wilmes P, Poetsch A, Dopson M. 2018. Multi-omics Reveals the Lifestyle of the Acidophilic, Mineral-Oxidizing Model Species Leptospirillum ferriphilumT. Applied and Environmental Microbiology, doi 10.1128/AEM.02091-17.
Johnson DB. 2014. Biomining—biotechnologies for extracting and recovering metals from ores and waste materials. Current Opinion in Biotechnology 30: 24–31.