04. Work Log
-
Genome assembly with Canu have been submitted to UPPMAX. Bach code that was submitted can be found in code/WGS/01_assembly_canu.sh. The six files provided in DNA_raw_reads where used as input.
-
Since time is limited we have been provided with trimmed reads for the full data set. However I will preform quality control and trimming on part of the data to try this part of the analysis. I have chosen to do this on one sample from continuous culture and one sample from bioleaching culture. The sample name and treatment can be seen below. I have run FastQC on these samples (forward and reverse). Code for this can be found in code/RNA/01_quality_raw_reads_fastqc.sh
| Sample Name | Treatment |
|---|---|
| ERR2036629 | Continous |
| ERR2117290 | Bioleaching |
- I have tried running things on UPPMAX both in interactive mode and by submitting a job to the queue!
-
The first genome assembly is done. I will spend some time today understanding the output from both Canu and FastQC which I ran yesterday for two of the samples. I will also check the quality of the assembly with QUAST and MUMmer.
-
QUAST was run on the assembly using the provided reference genome. The code for this analysis can be found in code/WGS/02_quality_quast.sh.
-
Assembled genome was aligned with the provided reference using nucmer. The code for this can be found in code/WGS/03_quality_align_nucmer.
-
I also started looking in to ways of plotting the reference genome and aligned contings and I think I found a nice R package.
- I've tried to really understand the output from all the different software. I feel like they are somewhat conflicting and I'm a little bit unsure on how to proceed. The N50 value is good (2563357) but there is some kind of missasembly. Both the output from QUAST and the alignment with MUMmerplot suggests a translocation (I think). The longest aligned block from both nucmer is ~1,4 mbp (see fig 1) but QUAST seems to align 97.566% (see fig 2) of the reference genome while this is not the case for nucmer (why??). I would like to try run Canu again but I need to read more about the different settings.
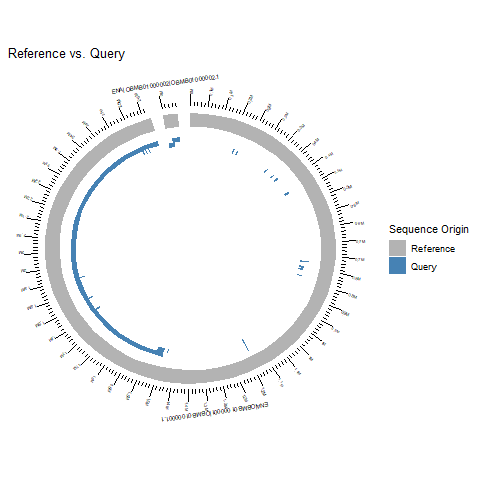
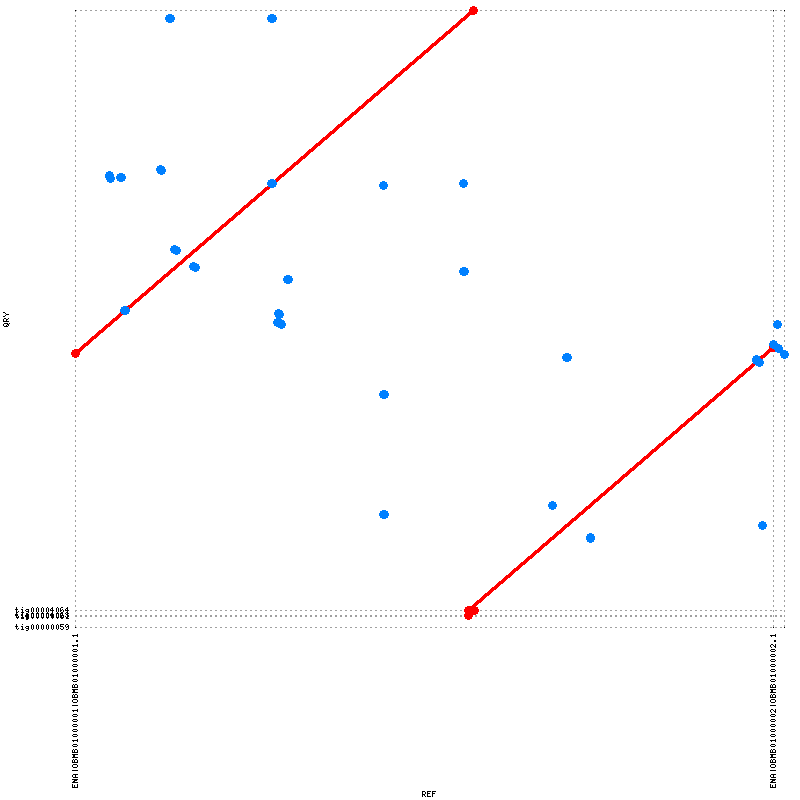
Figure 1. To the left: Alignment of reference genome and contigs (above 80% identity) by nucmer. To the right: Dot plot of the alignment (nucmer) between the contigs and the reference genome by MUMmerplot.
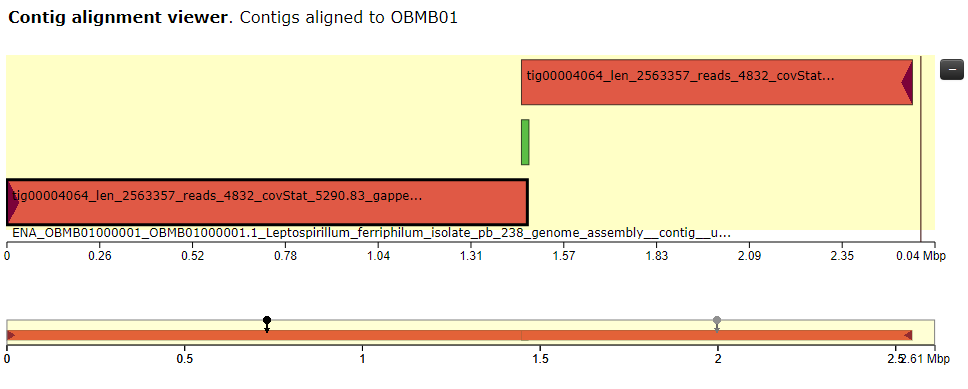
Figure 2. The aligned contigs based on QUAST. Two contigs aligned and the longer contig split into two because of the missasembly event (translocation).
- I'm running trimmomatic and fastQC on the trimmed reads. I thought it was a bit hard to know which parameters to use for trimmomatic but after reading a bit I think I have selected pretty standard values.
-
I have come to the conclusion that the cause of the weird alignment might be due to the fact that the genome is circular! Canu does not have specific parameters for circular genomes but this can be corrected for using tools like circulator. I will look into this... So I think I have come to the conclusion that it might not be necessary to rerun canu. It might be better to try to polish the output instead.
-
I'm running circlator on the canu assembly using the corrected reads from canu as well. It seems to be recommended to run som additional polishing steps after circlator using example quiver or arrow but I don't think any of these are available on uppmax. UPDATE: quiver/arrow is available I just didn't know the correct module name.
-
MultiQC is very nice!
- I've been sick and on Easter break but now I'm back and not to much behind. Circlator did not work last time I tried so I will try now again.
- I have given up on circlator. It is working but it is very very slow to run. I don't think its worth it even if it would have been interesting to see if it would have improved the assembly... I will move on to do some other assembly refinement.
- I will look in to arrow as a way of polishing my genome assembly.
- I'm running arrow!
- I'm going through the results from trimming the Illumina RNA-seq reads and trying to understand what everything means.
- Arrow is still running... I don't know if this is good or bad. I hope it works out and I get good results because I kind of need to move on. If it doesn't finish today I think I will proceed and then maybe I can get back to this if I have time later.
- I've prepared the scripts for annotation both with prokka and eggnog mapper and I will run them later today.
- I forgot to write here all last week but I'll update now what I have done.I have annotated the genome using both prokka and eggnogg (supplying the predicted genes from prodigal). I first tried to write a script to run eggnogg mapper but for some reason this didn't work so I decided to run in online instead. I have also worked on a script to compare the two annotations. I have also mapped the clean reads provided to the assembled genome. Next step will be to finish the "consensus" annotation and count the reads.
- In the last days I have finished a script that combines the annotations from prokka and eggnog. I have also started on mapping the RNA reads to the annotated genome.
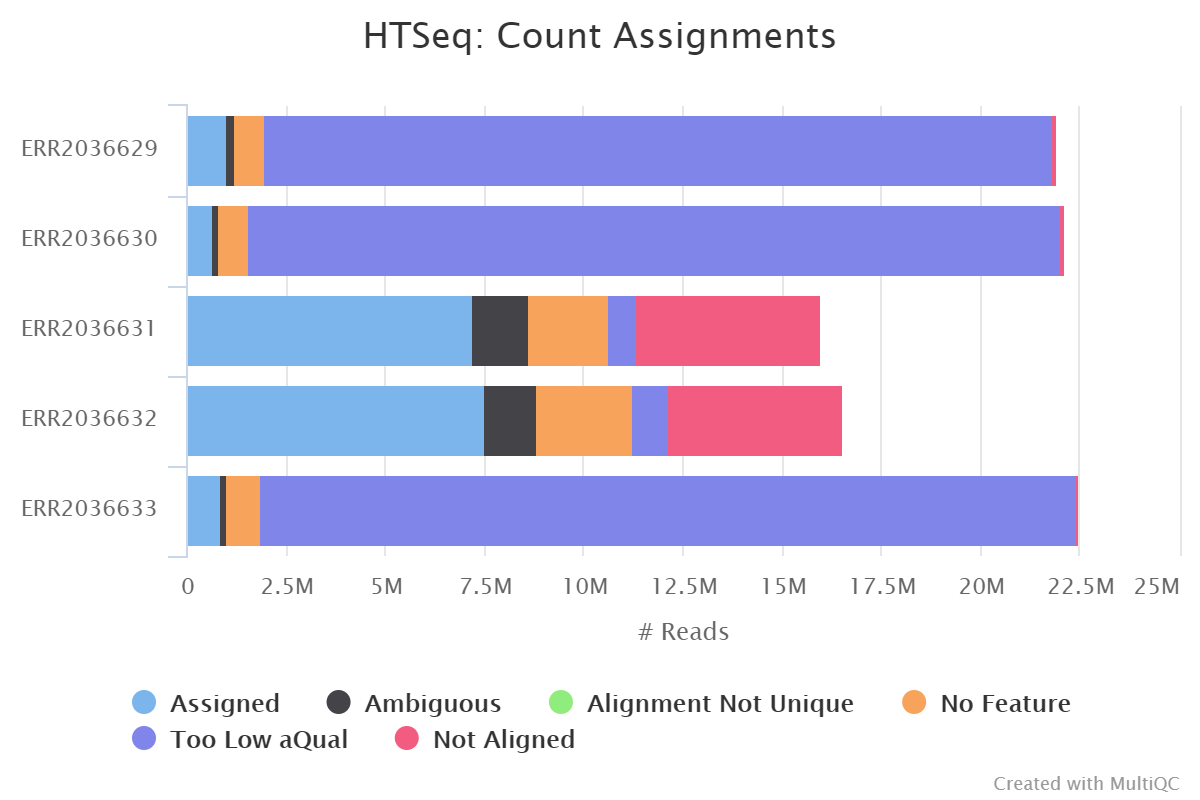
- I mapped the reads to the annotated genome and preformed differential gene expression analysis yesterday. However I thought that the counts produced by htseq where pretty low (48% of features had two low counts to be included in the analysis). I realized that -s should be "reverse" and not "yes" since illumina truseq stranded is dUTP based. I am running this again but right now I think rackham is down... I will focus on the synteny analysis and results from previous steps that I haven't included here yet.
- I think I'm doing ok right now in terms of time even though I've spent a lot of time on our other course the past week.
- So Uppmax was up again yesterday. I've rerun HTSeq with -s set to "reverse" which gave better results than the default value for -s (which is "yes"). Only 2% of features where discarded due to low counts compared to over 40% when running DESeq2. The percentage of up and down regulated genes where also a lot higher (~15% each) at p-val=0.1. I could probably use a lower p-value cutoff. However a very, very large amount of reads could not be mapped back to the genes when running HTSeq. For the continuous samples over 90% of reads did not map to a feature and mainly because of low quality alignment. Not sure what to do about this. Default which I used is 10 which I guess I could lower but I don't know if I have time...
- The past week when Uppmax has been down have mainly worked answering questions and working on scripts to run when uppmax is back.
- I have run synteny analysis with satsuma2 but I've run into trouble when trying to visualize the results. The software (MizBee) is not working on my computer.
- I reran htseq once more setting aQUal=0 just to check if this is the problem but this resulted in the majority of reads (over 90% for the contentious samples) being mapped to no feature which is really weird. I have visualized the genome and the predicted genes and the reads in IGV and which looks fine to me so I'm not really sure what the problem is. It's strange that there is such a difference between the two treatments and I would not trust the downstream analysis based on this. I've spent the morning today trying to figure out the reason for such a low percentage of reads mapping to my features but I cant find anything out...
- After discussion with the TAs it seems like my results are not so weird and probably mostly due to rRNA which exist in a large number of copies in the DNA. This gives them a low quality score and would also explain why they show up as no feature when I lowered the threshold for this. At least my results improved significantly when I changed from --stranded=yes to --stranded=reverse so this investigation into the low counts is not completely wasted (se below... "yes" to the top and "reverse" to the bottom)!
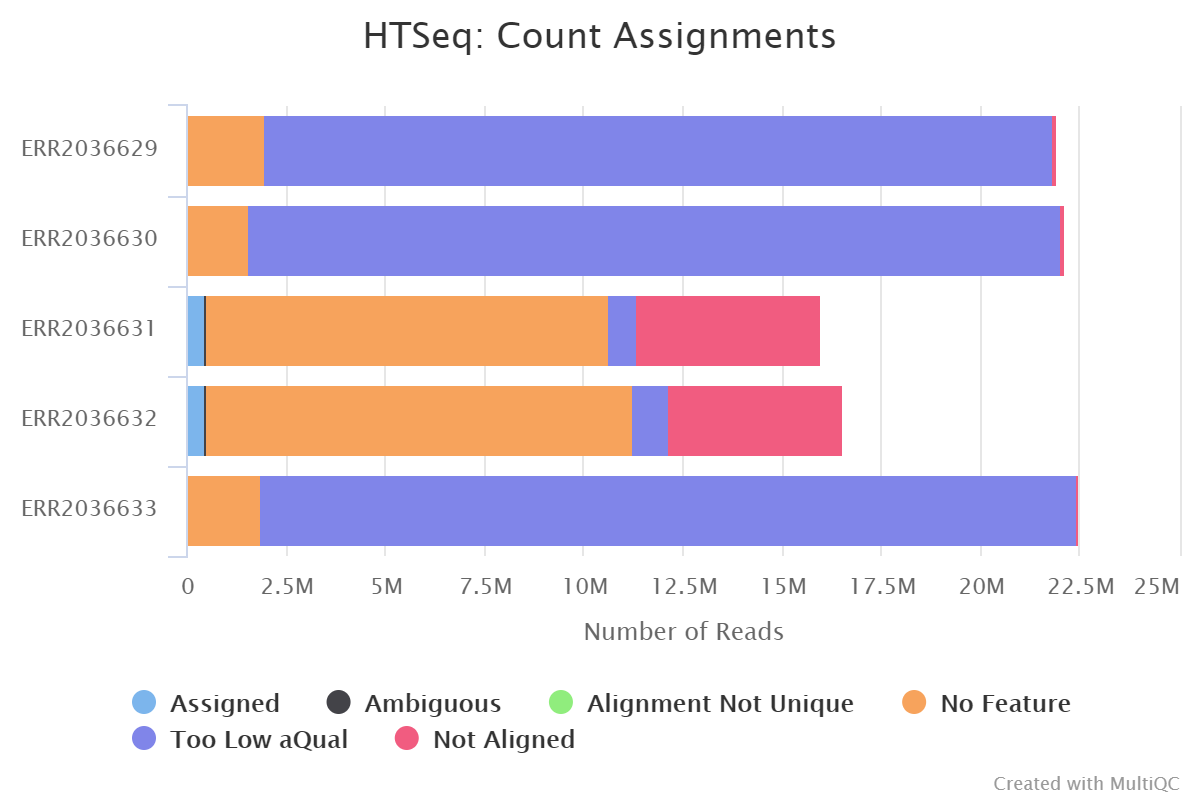
- The last week I finished all the basic analysis. The past few days I've been working on completing the wiki, making the presentation and doing some extra stuff such as promoter prediction etc.