Taxonomy alignment tools
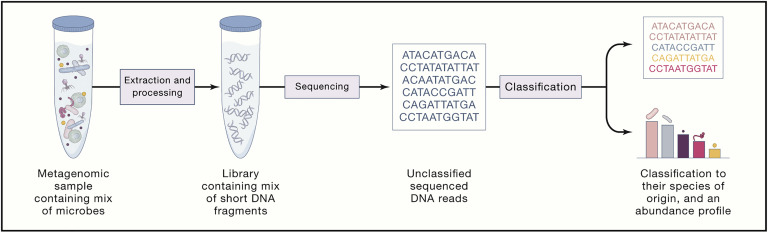
Many bioinformatics tools have been developed to classify the taxonomic profile of metagenomic data. Each tool uses different approaches and databases. Thus, it is important to choose the right tools based on the sample type (i.e., some tools were designed only for human metagenomics samples). A few benchmarking papers can be referenced and help you decide which tool would be best for your interest. Here, we are comparing multiple tools regarding their usage and results. Note that we are not recommending certain tools superior to others.
Resrouces:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1646-y
https://www.sciencedirect.com/science/article/pii/S0092867419307755
https://www.nature.com/articles/nmeth.4458
Methods for analyzing shotgun sequencing can be placed into three broad categories: assembly-based, alignment-free, and alignment-based. Assembly-based approaches require merging overlapping DNA fragments into longer sequences which can be assembled into whole genomes. However, this is not always possible. Alignment-free methods involve the DNA sequences themselves, such as matches of k-mers between reference genomes and the DNA sequences within a sample. This approach has shown to be produce a high number of misclassifications and false positives. Alignment-based methods, on the other hand, require the alignment of DNA fragments to previously characterized reference sequences using alignment algorithms. Many alignment tools exist, but it has been shown that MALT (MEGAN Alignment tool) outperforms the other ones.
MALT is an alignment-based tool that allows researchers to query DNA sequences against reference databases. MALT can align nucleotide sequences to nucleotide databases (MALTn) or nucleotide to amino acid databases (MALTx).
In this section, you will be given the code that is used to index the reference database, align sequences to the database, and convert these into RMA files. RMA files store reads, alignments, as well as taxonomic and functional classification information for a given sample. The file is indexed so that it is easy to access the reads and alignments.
Index the reference database
malt-build -i RefSeqONLY-Assembly-Arch-Bact-Genome-Chromosome-Scaffold-27-7-17/CATTED-arch-bact.fna.gz -d MALT_BUILD \
--sequenceType DNA > malt-build.log
Make sure the malt environment is active
source env_malt_038.sh
# set variables
# directories where you store the samples
SAMPLE_FOLDER=Samples
# path where you store the database
DATABASE=/store/common/data/MALT_BUILD_v_0.6.1
# save the RMA output files to a specific folder
mkdir RMA_FILES
RMA=RMA_FILES
# save the BLAST output files to a specific folder
mkdir BLAST_FILES
BLAST_FILES=BLAST_FILES
# begin malt-run
malt-run -i $SAMPLE_FOLDER/P1_PortugeseKing_trimmed_merged.fastq -d $DATABASE --mode BlastN -at SemiGlobal --minPercentIdentity 90 --minSupport 0 --maxAlignmentsPerQuery 25 --gapOpen 7 --gapExtend 3 --band 4 -o RMA_FILES --memoryMode load --verbose true -t 40 --alignments RMA_FILES --format Text
malt-run -i $SAMPLE_FOLDER/P2_PortugeseKing_trimmed_merged.fastq -d $DATABASE --mode BlastN -at SemiGlobal --minPercentIdentity 90 --minSupport 0 --maxAlignmentsPerQuery 25 --gapOpen 7 --gapExtend 3 --band 4 -o RMA_FILES --memoryMode load --verbose true -t 40 --alignments RMA_FILES --format Text
-m --mode; legal values include BlastN, BlastP, BlastX
-at --alignmentType; type of alignment to be performed. Default value is Local. Legal values include Local, SemiGlobal.
--minPercentIdentity; minimum percent identity used by LCA algorithm (default: 0)
--minSupport; minimum support value for LCA algorithm
--maxAlignmentsPerQuery; maximum alignments per query (default: 25)
--gapOpen; gap open penalty (default: 11)
--gapExtend; gap extension penalty (default: 1)
--band; band width for banded alignment (default: 4)
-o; output RMA files
--memoryMode; memory mode (default: load, page, map)
--verbose; read out each command line option (default: false)
--alignments; output alignment files
--format; output alignment format (default: SAM), legal values: SAM, Tab, Text
blast2rma is similar to the sam2rma program as it generates RMA files from BLAST output.
blast2rma -i $RMA/P1_PortugeseKing.blastn -r $SAMPLE_FOLDER/P1_PortugeseKing_trimmed_merged.fastq -o $BLAST_FILES/P1_PortugeseKing.blast2rma -ms 44 -me 0.01 -f BlastText -supp 0.1 -alg weighted -lcp 80 -t 40 -v --blastMode BlastN
blast2rma -i $RMA/P2_PortugeseKing.blastn -r $SAMPLE_FOLDER/P2_PortugeseKing_trimmed_merged.fastq -o $BLAST_FILES/P1_PortugeseKing.blast2rma -ms 44 -me 0.01 -f BlastText -supp 0.1 -alg weighted -lcp 80 -t 40 -v --blastMode BlastN
-i; input BLAST files
-r; reads file(s) (fasta or fastq)
-o; output file
-ms, --minScore; Min score (default: 50.0)
-me, --maxExpected; Max expected (default: 0.01)
-f; format, input file format
-supp, --minSupportPercent; min support as percet of assigned reads (default: 0.01)
-alg, --lcaAlgorithm; set the LCA algorithm to use for taxonomic assignment, legal values: naive, weighted, longReads
-lcp, --lcaCoveragePercent; set thhe percent for the LCA to cover (default: 100.0)
-t; threads
-v; verbose commands
--blastMode; legal values BlastN, BlastP, BlastX, Classifier
nohup cat Samples.txt | while read samples; do malt-run -i $samples -d $DATABASE --mode BlastN -at SemiGlobal --minPercentIdentity 90 --minSupport 0 --maxAlignmentsPerQuery 25 --gapOpen 7 --gapExtend 3 --band 4 -o RMA_FILES --memoryMode load --verbose true -t 40 --alignments RMA_FILES --format Text ; blast2rma -i $RMA/$samples.blastn.gz -r $samples -o $BLAST_FILES/$samples.blast2rma -ms 44 -me 0.01 -f BlastText -supp 0.1 -alg weighted -lcp 80 -t 40 -v --blastMode BlastN ; done
Kraken 2 is the newest version of Kraken, a taxonomic classification system using exact k-mer matches to achieve high accuracy and fast classification speeds. This classifier matches each k-mer within a query sequence to the lowest common ancestor (LCA) of all genomes containing the given k-mer. The k-mer assignments inform the classification algorithm.
Bracken (Bayesian Reestimation of Abundance with KrakEN) is a highly accurate statistical method that computes the abundance of species in DNA sequences from a metagenomics sample. Braken uses the taxonomy labels assigned by Kraken, a highly accurate metagenomics classification algorithm, to estimate the number of reads originating from each species present in a sample. Kraken classifies reads to the best matching location in the taxonomic tree, but does not estimate abundances of species. We use the Kraken database itself to derive probabilities that describe how much sequence from each genome is identical to other genomes in the database, and combine this information with the assignments for a particular sample to estimate abundance at the species level, the genus level, or above. Combined with the Kraken classifier, Bracken produces accurate species- and genus-level abundance estimates even when a sample contains two or more near-identical species.
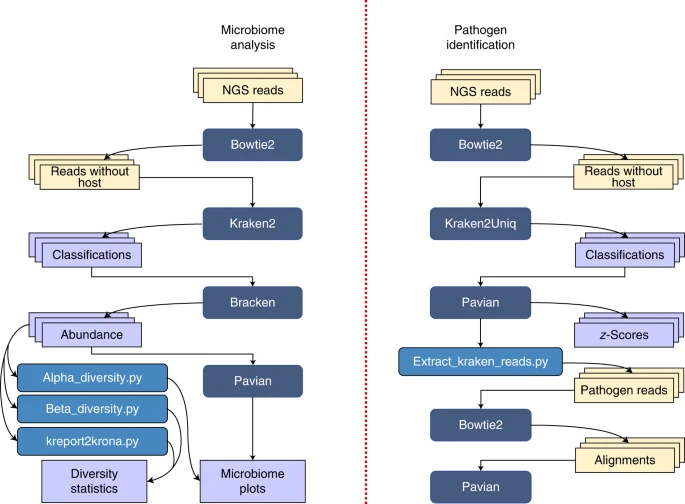
NOTE: This tutorial actively mirrored their official published protocol

Installation
mamba install -c bioconda kraken2
mamba install -c bioconda bracken
Database
One of the disadvantage of using Kraken2 is their huge size of database. They also allow to use custom database which could be smaller (but not complete), or also provide pre-built database. Several prebuilt Kraken 2 databases are available at https://benlangmead.github.io/aws-indexes/k2. The most commonly used database is the standard Kraken 2 database (which includes RefSeq archaea, bacteria, viruses, plasmid complete genomes, UniVec Core and the most recent human reference genome, GRCh38).
Use pre-build database (recommended)
- Go to https://benlangmead.github.io/aws-indexes/k2
- Right click on database you desired (HTTPS URL), click "Copy link"
- Download database using wget in your command line and unzip
mkdir kraken2_database # Create folders for database
cd kraken2_database # change directory to kraken2_database
wget https://genome-idx.s3.amazonaws.com/kraken/k2_standard_20221209.tar.gz # Download standard database
tar -xzvf k2_standard_20221209.tar.gz # Decompress your database
If you want to build a standard/custom database (Refseq complete genome database), you can create Kraken2 database
kraken2-build --standard --db <desired db name>
OR if you want to build one or more reference libraries
kraken2-build --download-taxonomy --db <desired db name>
kraken2-build --download-library bacteria --db <desired db name>
Classification
Run Kraken2 to classify all reads in each sample
kraken2 --db kraken2_database <sample_pair1.fastq file> <sample_pair2.fastq.file> --report <FILENAME.report>
--output <FILENAME> --paired --classified-out <FILENAME_CLASSIFIED> --threads 20
--output : output file name as FILENAME
--paired : input files have paired-end reads
--classified-out : print classified sequences to FILENAME_CLASSIFIED
You will get two ouput files from Kraken2
.kraken2 # a standard Kraken2 output
.k2report # a Kraken2 report file
Bracken
run bracken for abundance estimation of microbiome samples
bracken -d kraken2_database -i <.k2report file from the previous step> -r <read_length>
-l <taxonomic_level> -o BRACKEN_OUTPUT -t NUM_THREADS
-d: name of database
-i: input kraken2 report file
-r: read length (i.e., 150 or 250)
-l: taxonomic level [Default = 'S', Options = 'D','P','C','O','F','G','S']
-o: Bracken_output prefix
-t: number of threads
Results report
Check example Kraken2/Bracken output files
Create biom file from multiple Kraken2/Bracken reports
mamba install -c bioconda kraken-biom ## Install kraken-biom
kraken-biom S1.report S2.report S3.report --max P --min S ## change the max and min taxonomic level as phylum(P) and species(S)
mOTU is a computational tool that estimates relative abundance of known and unknown microbial community members using metagenomic shotgun data.
One of the notable advantage of using mOTU is that they are using extended amount of data not just from complete genome sequences, but also metagenomic samples and meta genome assembled genomes (MAGs). On the other hands, they are extracting marker genes and mOTUs (~species) contained in the mOTUs database, meaning that they are extracting marker gene sequences from the entire database and apply for the classification.
Reference : https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-022-01410-z
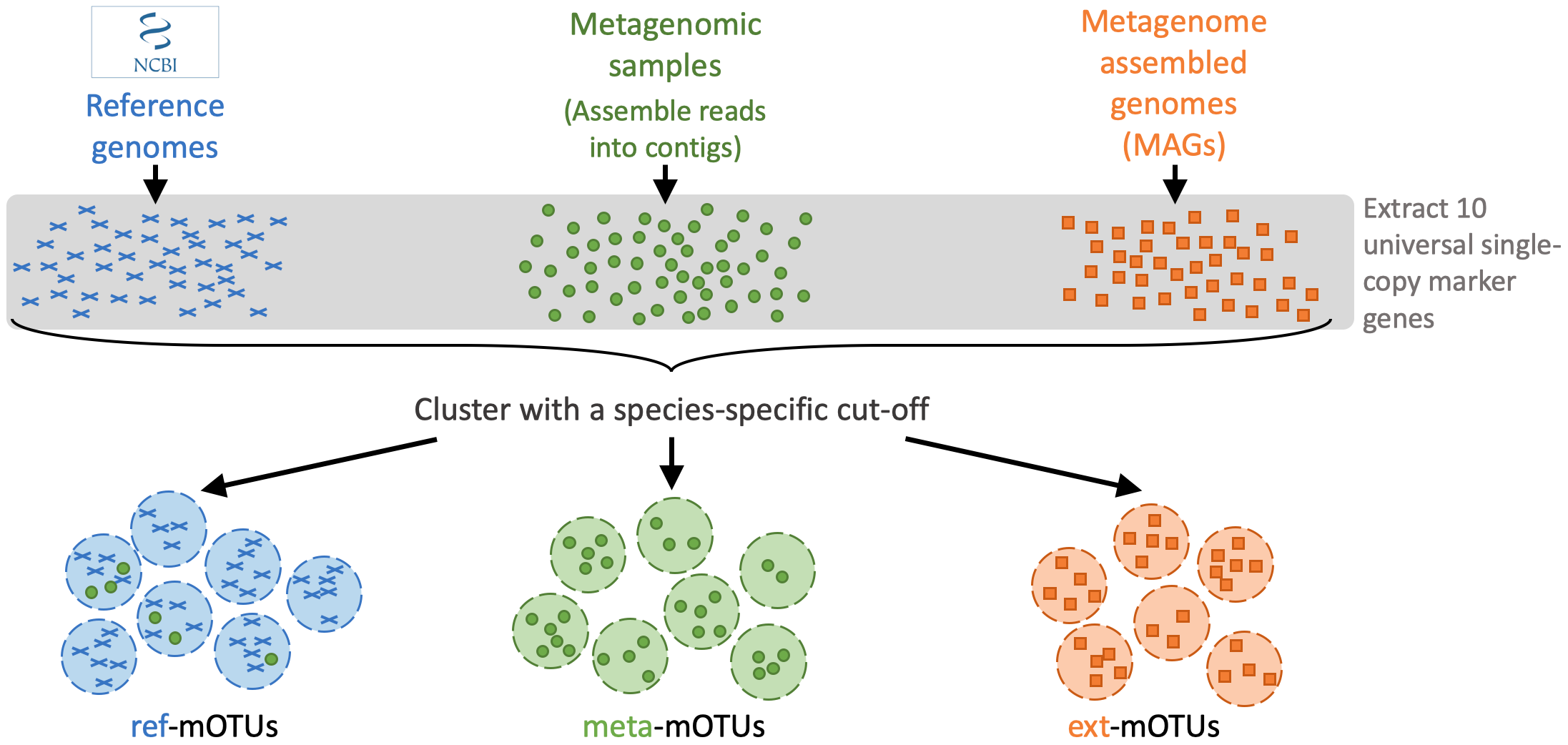
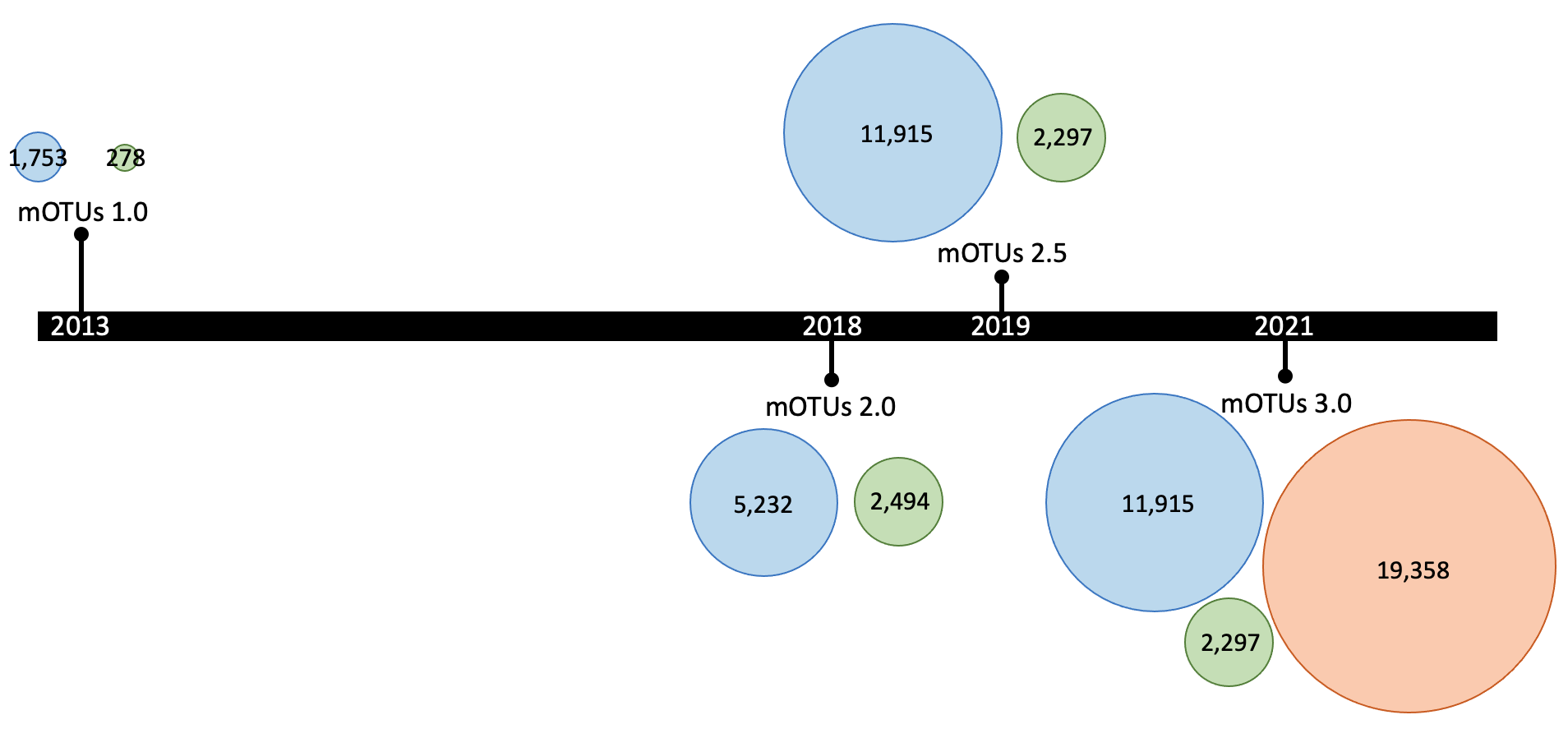
A mOTUs database is composed of three types of mOTUs:
ref-mOTUs, which represent known species, meta-mOTUs, which represent unknown species obtained from metagenomic samples, ext-mOTUs, which represent unknown species obtained from MAGs.
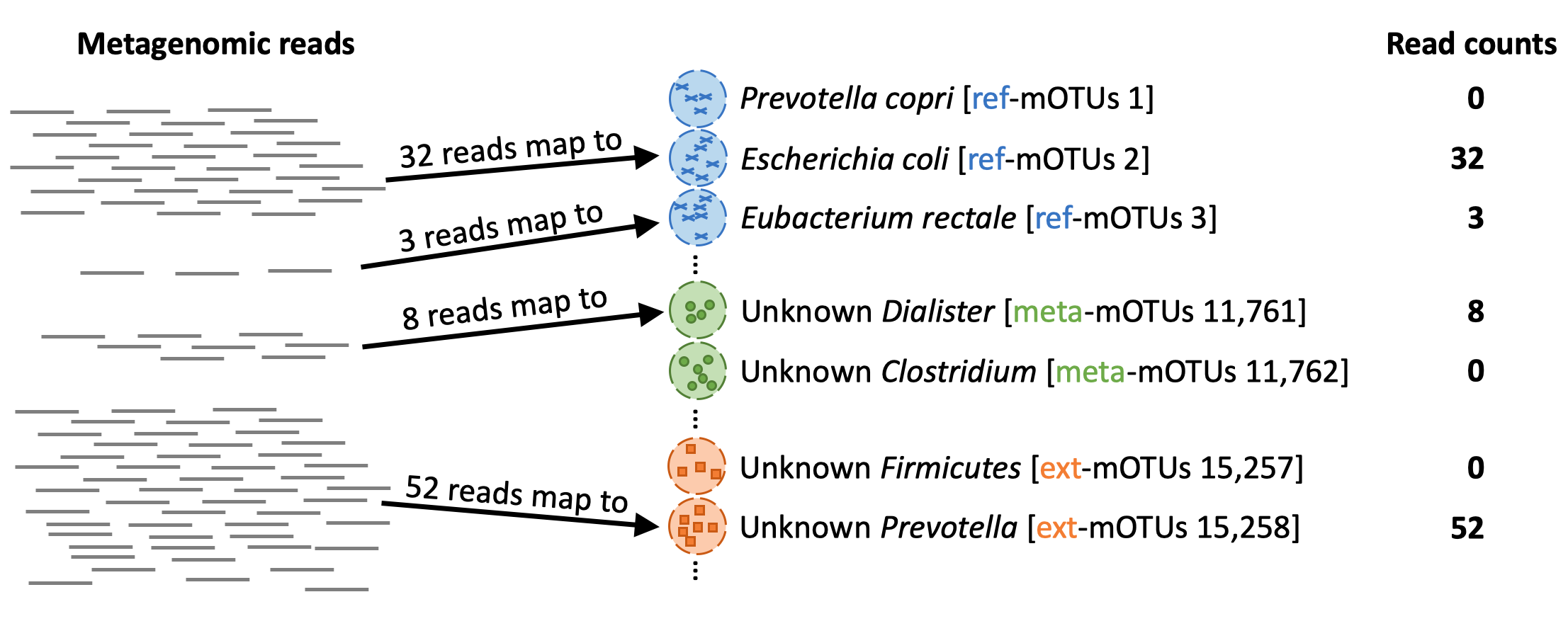
When profiling a meta gnomic sample, each reads will be mapped to the genes in the different mOTUs (marker genes)
Installation
mamba install -c bioconda motus
# Download the mOTUs database (~3.5GB)
motus downloadDB
Classification
Classification for one sample
## Automated pipeline from raw reads to profile
motus profile -f [forward fastq file] -r [reverse fastq file] -t [number of threads] > [output file name.txt]
## Independent steps
motus map_tax -f [forward fastq file] -r [reverse fastq file] -o [output .sam file]
motus calc_mgc -i [output .sam file from the previous step] -o [output .count file]
motus calc_motu -i [output .count file from the previous step] > [output file name as .txt]
## Merge multiple runs into the one file
motus profile -f [forward fastq file 1] -r [reverse fastq file 1] -t [number of threads] > [output file name .txt 1]
motus profile -f [forward fastq file 2] -r [reverse fastq file 2] -t [number of threads] > [output file name .txt 2]
motus profile -f [forward fastq file 3] -r [reverse fastq file 3] -t [number of threads] > [output file name .txt 3]
motus merge -i [output file name .txt 1],[output file name .txt 2],[output file name .txt 2] > all_sample_profiles.txt
OR
You can profile multiple samples at once
motus profile -f sample1_run1_for.fastq,sample1_run2_for.fastq -r sample1_run1_rev.fastq,sample1_run2_rev.fastq -s sample1_run1_single.fastq > taxonomy_profile.txt
Results
Check example mOTUs output files
MetaPhlAn relies on unique clade-specific marker genes identified from ~1M microbial genomes. The latest version of MetaPhlAn (MetaPhlAn4) is based on 1.01M prokaryotic reference and MAGs.
For more information, please read the original paper of MetaPhlAn4 - https://www.nature.com/articles/s41587-023-01688-w
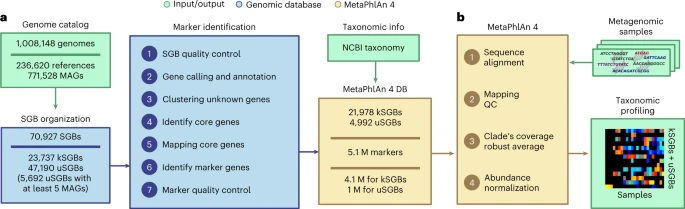
Blanco-Miguez et al., 2023, Figure 1.: MetaPhlAn 4 integrates reference sequences from isolate and metagenome-assembled genomes for metagenome taxonomic profiling. a, From a collection of 1.01 M bacterial and archeal reference genomes and metagenomic-assembled genomes (MAGs) spanning 70,927 species-level genome bins (SGBs), our pipeline defined 5.1 M unique SGB-specific marker genes that are used by MetaPhlAn 4 (avg., 189 ± 34 per SGB). b, The expanded marker database allows MetaPhlAn 4 to detect the presence and estimate the relative abundance of 26,970 SGBs, 4,992 of which are candidate species without reference sequences (uSGBs) defined by at least five MAGs. The profiling is performed firstly by (1) aligning the reads of input metagenomes against the markers database, then (2) discarding low-quality alignments and (3) calculating the robust average coverage of the markers in each SGB that (4) are normalized across SGBs to report the SGB relative abundances (see Methods). All data are presented as mean ± s.d.
Installation
mamba install -c conda-forge -c bioconda metaphlan
## Install database
metaphlan --install --bowtie2db [database installation folder]
Classification
Important! The MetaPhlAn 4 database has been substantially increased in comparison with the previous 3.1 version. Thus, for running MetaPhlAn 4, a minimum of 15GB or memory is needed.
metaphlan [forward_reads.fastq,reverse_reads.fastq] --bowtie2out output.bowtie2.bz2
--nproc 5 --input_type fastq --unclassified_estimation -o profiled_metagenome.txt
--bowtie2db [installed database folder]
--nproc : Number of processor
--input_type : type of sample extension
--unclassified_estimation : Estimate unclassified reads
-o: output file name
--bowtie2db: Installed database location from the database installation step
Merge multiple tables into one
merge_metaphlan_tables.py metaphlan_output1.txt metaphlan_output2.txt metaphlan_output3.txt > output/merged_abundance_table.txt
Results
TBD